





Abstract
Rapid response to the current coronavirus disease 2019 (COVID-19) pandemic requires fast dissemination of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) genomic sequence data in order to align diagnostic tests and vaccines with the natural evolution of the virus as it spreads through the world. To facilitate this, the National Library of Medicine’s National Center for Biotechnology Information developed an automated pipeline for the deposition and quick processing of SARS-CoV-2 genome assemblies into GenBank for the user community. The pipeline ensures the collection of contextual information about the virus source, assesses sequence quality and annotates descriptive biological features, such as protein-coding regions and mature peptides. The process promotes standardized nomenclature and creates and publishes fully processed GenBank files within minutes of deposition. The software has processed and published 982 454 annotated SARS-CoV-2 sequences, as of 21 October 2021. This development addresses the needs of the scientific community as the sequencing of SARS-CoV-2 genomes increases and will facilitate unrestricted access to and usability of SARS-CoV-2 genomic sequence data, providing important reagents for scientific and public health activities in response to the COVID-19 pandemic. Database URL https://submit.ncbi.nlm.nih.gov/sarscov2/genbank/
Introduction
The coronavirus disease 2019 (COVID-19) pandemic caused by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) is a global health concern that has disrupted economies and personal lives, causing sickness and loss of life. Research around the world is focused on developing and improving diagnostic tools and vaccines for SARS-CoV-2 and tracking and understanding the evolution of the virus. This relies on the availability of high-quality nucleic acid sequences as a basis for discovery (1–5). The importance of making global SARS-CoV-2 sequence data quickly and readily accessible with rich contextual detail, describing where and when the virus was isolated from samples across the world, in a central, unrestricted and familiar repository cannot be overemphasized.
In response to this need, we developed an automated SARS-CoV-2 submission pipeline to expedite the publication of SARS-CoV-2 sequence data to the GenBank database. GenBank is the public comprehensive genetic sequence database (6) housed at the National Library of Medicine’s (NLM’s) National Center for Biotechnology Information (NCBI), which relies on the input of genetic sequence data from the international scientific research communities. GenBank is freely available via an internet connection, its access is unrestricted and it is widely accessed and cited by a diversity of users. GenBank, together with the International Nucleotide Sequence Database Collaboration (INSDC) partners DNA Data Bank of Japan (DDBJ) and European Nucleotide Archive (ENA), is designed to provide and encourage access within the scientific community to the most up-to-date and comprehensive DNA sequence information (7).
Input of data into GenBank relies on users to provide their data through a webpage or file deposition. Given the all-inclusive nature of the sequence types accepted to GenBank, GenBank submission tools have generally been built with flexibility in mind, offering access to the wide range of potential features appropriate to different sequence types. The submission tools have allowed for variability in biological data, while also evaluating data accuracy and ensuring data conforms to database formatting standardizations, with the end goal of adhering to FAIR (Findable, Accessible, Interoperable, and Reusable) data principles (8). Additionally, the tools must be accessible and usable to a wide range of users with varying levels of expertise. This flexibility and inclusivity have made it difficult for some users to figure out what was needed and appropriate for their own submission.
One way we have begun to address this complexity and expedite public release of sequences critical to public health, as well as other commonly submitted sequence types, is by creating specialized GenBank sequence submission web-based wizards. Each wizard is focused on a specific sequence type and presents only options relevant to that type. Additionally, an FTP-based submission method has been developed for data deposition for users who wish to submit as part of a computational pipeline. Subsequent processing of deposited data received via web and FTP is automated to facilitate rapid data release. During processing, sequences are analysed and validated, annotation is added for features such as proteins and mature peptides, accession numbers are assigned and data are released to the public databases. Here, we present the SARS-CoV-2 GenBank submission wizard. The requirements, validations and processing steps for this wizard will be discussed. Sequence submissions that pass validations through this pipeline are accessible from the NLM/NCBI SARS-CoV-2 resources page (https://www.ncbi.nlm.nih.gov/sars-cov-2/), which catalogues all COVID-19-related data available from NLM/NCBI including sequences, literature and clinical trials.
Materials and methods
The SARS-CoV-2 submission pipeline is designed to accept, validate and annotate partial and complete genome sequences for publication to GenBank. The information in this section outlines the files and information a submitter would use to submit SARS-CoV-2 sequence data and is followed by a description of the processing workflow implemented by NLM/NCBI.
Successful entry into the full processing pipeline requires a login, input of data and files in web-based forms or providing all required data in files via FTP. There are no restrictions for either method; however, submitters with a larger number of sequences may find the deposition of data by FTP easier.
In the web-based submission wizard (https://submit.ncbi.nlm.nih.gov/sarscov2/genbank/), a data submitter is guided through a series of pages for providing the pertinent information to complete the submission (Figure 1). The following information is collected: contact information for the authors, sequencing technology and assembly methodology used to generate the data, sequence data in FASTA format, a minimum set of source metadata either as tab-delimited table or provided through input forms, sequence authors and reference information (if applicable). Related BioProject, BioSample and Sequence Read Archive (SRA) accession numbers are optional but may also be provided together with source metadata. Biological sequence features, such as genes, coding regions, mature peptides and stem-loops, are automatically added after the user submits (during processing at NLM/NCBI), so this information does not need to be provided by the submitter.
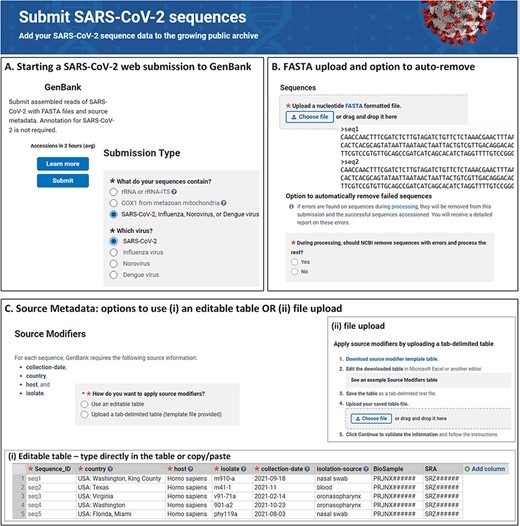
NLM/NCBI SARS-CoV-2 web submission forms for starting a submission (A), uploading sequences (B) and providing source metadata (C). Screenshots are from https://submit.ncbi.nlm.nih.gov/sarscov2/ and https://submit.ncbi.nlm.nih.gov/subs/genbank/.
As an alternative to data entry in web-based forms, input files may be provided via FTP. The following information is required: FTP account, sequence data in FASTA format, tab-delimited source metadata including at least organism, isolate, host, collection date and country and a submission template that contains contact information as well as sequencing and assembly information. Additional optional information may be provided. Submission templates can be created using https://submit.ncbi.nlm.nih.gov/genbank/template/submission/. More extensive documentation is available for SARS-CoV-2 FTP-based submission to NLM/NCBI (https://submit.ncbi.nlm.nih.gov/sarscov2/genbank/).
Upon notification that a submitter has made a SARS-CoV-2 submission, compute resources at NLM/NCBI process the submission data through a series of automated steps linked together through the Apache Airflow workflow framework. The workflow trims ambiguous sequence ends, removes vector, checks the sequences for minimum and maximum length requirements, performs taxonomy lookups, validates and annotates sequences using the Viral Annotation DefineR (VADR) software package (9), executes basic clean-ups, sorts errors and loads the data to the public database or triggers submission correction (workflow outlined in Figure 2).
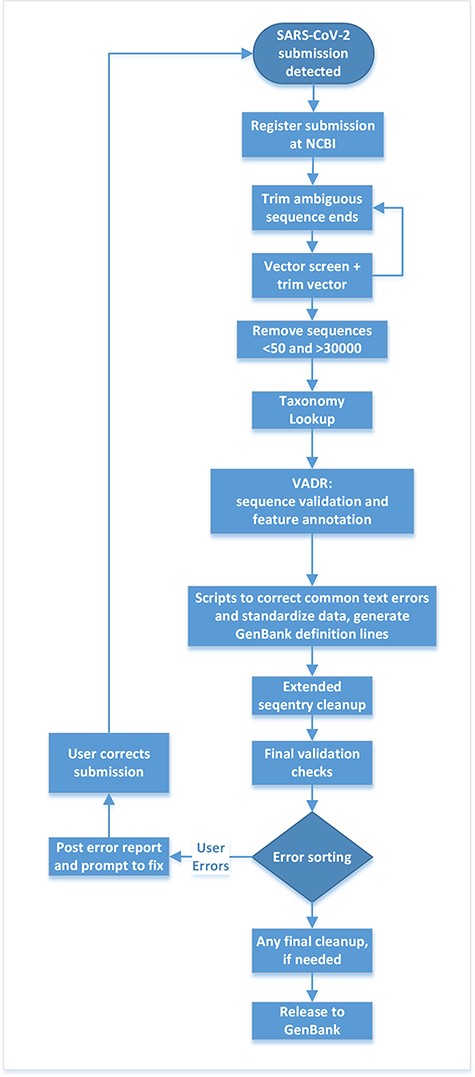
Post-submission processing steps performed at NLM/NCBI to evaluate, annotate and prepare SARS-CoV-2 sequence data for public release to the GenBank database.
Results and discussion
Source metadata processing and validation for SARS-CoV-2 submissions to GenBank
For source metadata, a minimal set of requirements and standards were established through outreach and collaboration with the SPHERES consortium (Sequencing for Public Health Emergency Response, Epidemiology, and Surveillance; https://www.cdc.gov/coronavirus/2019-ncov/variants/spheres.html) and PHA4GE working group (https://pha4ge.org/). Establishing these minimal requirements for source metadata was done to make the data useful for comparative analysis, research discovery, trend monitoring and future data reuse. The required fields (known as source modifiers at GenBank) include isolate, collection date, locality and host and are collected to assess the WHO, WHAT, HOW, WHERE and WHEN descriptions of the samples. Examples for each field in the preferred format are provided as a guide for the user. For the isolate source modifier, in order to promote consistency and comparability in source metadata, the International Committee on Taxonomy of Viruses-recommended isolate format (SARS-CoV-2/host/location/laboratory ID/date) was adopted for GenBank submissions (10). Rather than requiring the user to supply the properly formatted isolate and risking variation in this standardized field, the submission tool constructs a well-formatted isolate based on the laboratory ID provided in this field and other required source metadata. A valid collection date is required and is automatically converted to the ISO format. The country field is automatically checked and standardized to formats agreed upon by the INSDC (e.g. USA: Maryland). Information about the host organism in which the SARS-CoV-2 sample was found is required and stored in the ‘host’ source modifier. Additional source metadata fields may be provided with the GenBank submissions, such as isolation source, which describes details about the physical environment of the biological sample from which the sequence was derived. A list of other source modifiers that may be provided is available (https://www.insdc.org/files/feature_table.html).
Common variations in user-provided source modifier values are detected and automatic conversions to a standardized format are made to help ease the burden on the submitter. For example, common host typos for Homo sapiens are detected and corrected. There are cases, however, where a value is deemed invalid by the software since it is not close enough to the expected format. Invalid or missing values for these required source modifier fields trigger an error for the submitting user to fix and respond to, enforcing richer context for the sequence data for all users of the database.
Submitters are encouraged to submit the associated raw reads to SRA (https://submit.ncbi.nlm.nih.gov/subs/sra/). During this process, a BioProject, which describes the initiative, and a BioSample, which describes the physical sample, are also created. The three assigned accession numbers from BioProject, BioSample and SRA should be included in the GenBank assembly submission to provide linkage between these data. In most cases, the ‘SARS-CoV-2-clinical’ BioSample package should be used as it contains additional rich contextual metadata fields to describe information surrounding the infection, disease status and vaccination and treatment status (https://submit.ncbi.nlm.nih.gov/biosample/template/?organism-organism_name=&organism-taxonomy_id=&package-0=SARS-CoV-2.cl.1.0&action=definition). Templates for non-host-derived samples, such as SARS-CoV-2 wastewater samples, are also available to capture rich information in surveillance efforts to understand the reach of infection more fully.
Sequence processing and biological feature annotation
Several sequence checks and edits are performed to help ensure high quality data (Figure 2). This includes the following: trim terminal ambiguous nucleotides, remove sequences with >50% dispersed ambiguities, trim terminal vector and laboratory adaptor sequences using NLM/NCBI VecScreen (https://www.ncbi.nlm.nih.gov/tools/vecscreen/), remove sequences longer than 30 000 nucleotides and remove sequences shorter than 50 nucleotides. Long sequences are removed because the genome size of SARS-CoV-2 is less than 30 000 nucleotides and sequences longer than the expected genome size are indicative of problematic data. A list of edits performed and sequences removed is provided to the user either in the web wizard or in a report post-submission, depending on the submission size and method.
Feature annotation is performed automatically to make submission easier and to promote consistent annotation. GenBank uses the VADR software package (9; Nawrocki et al., manuscript in preparation) to align submitted sequences to reference SARS-CoV-2 genomes and to annotate stem-loop, coding region (CDS) and mature peptide features in the sequences based on that alignment and the existing reference genome annotation. VADR reports potential problems with each sequence as alerts, including regions of low sequence similarity to the reference and potential frameshift mutations in coding regions. These alerts could indicate new variants that do not align properly with existing references, or they could indicate problems with the sequence such as incorrect sequence assembly or low-quality sequence. If problems are detected, the data submitter is alerted about the problem and given the opportunity to upload a corrected file for reanalysis. If the data submitter has evidence the sequence is correct and there is a naturally occurring variation (for example, there is high sequence read coverage and/or experimental evidence is provided in support of a variation that led to a VADR alert), the submitter can communicate with NLM/NCBI to verify and allow release of the data. The VADR reference library will be updated to include additional references when new variants become widespread; for example, an alpha (B.1.1.7) reference sequence was added in January 2021. Information about the alerts generated by VADR as well as other test tools used by GenBank can be found at https://www.ncbi.nlm.nih.gov/genbank/sequencecheck/virus/ and https://www.ncbi.nlm.nih.gov/genbank/validation/. VADR can be downloaded and run locally by submitters prior to submission to determine if their sequences will generate alerts at this stage of the submission pipeline (https://github.com/ncbi/vadr/wiki/Coronavirus-annotation).
Feedback provided to the user
The web-based forms provide real-time validation and a preview of records generated from the information provided at the time of submission. A limited set of validations allows for immediate correction of data in the forms before the submission is completed (Figure 3). A data summary and preview of records are provided on the final page of a web submission (Figure 4), which provides the submitter an opportunity to see their data in the familiar GenBank format and correct typos or information they notice is incorrect before submitting. Complete validation, information about processing, comprehensive error reporting, annotated records and accession numbers matched to sequence_IDs are typically available for the submitter within 1–2 h after submission (Figure 5). Upon accession assignment, sequence records are immediately loaded to the public GenBank database unless the user has opted to hold the data private until a given date. The GenBank records are annotated with gene, stem-loop and coding region features as well as the conceptual protein translation and mature peptide features. Data with errors (including VADR alerts) are immediately presented to the submitter with explanations. Both web-based forms and the file deposition-based submission method have an option to either prioritize processing of sequences that pass all checks or hold the entire submission until it is determined to be error-free. The option to automatically remove sequences with errors is available for multiple sequences and results in faster accession assignment and rapid final processing of passing sequences, while sequences with VADR alerts are removed and reported back to the user for further review. If the option to auto-remove is declined by the user and there are sequences with errors, the system will report back with an error report and the opportunity to correct the data in the submission before accessions are assigned. While many errors are presented back to the user for correction, a minimal set of errors are subject to NLM/NCBI review and further processing.
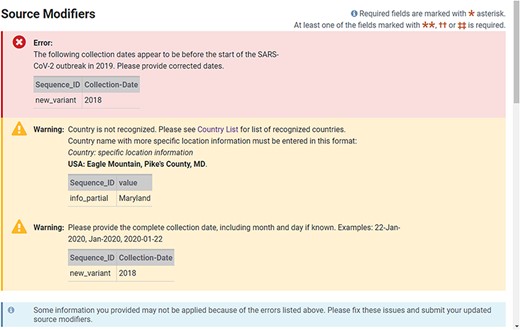
Example of real-time validation of source metadata during the web submission process prompting for immediate feedback and correction.
Submission statistics for sequences published by the NLM/NCBI SARS-CoV-2 GenBank processing pipeline, as of 21 October 2021
| Number of sequences with source metadata present | Number of sequences with data linkages (optional) | ||||||
|---|---|---|---|---|---|---|---|
| Total number of sequences published | Isolate | Country | Collection date | Host | Isolation source (optional) | Biosample linkage present | SRA linkage present |
| 982 454 | 982 451 (99.9%) | 982 438 (99.9%) | 982 418 (99.9%) | 982 379 (99.9%) | 547 014 (55.7%) | 245 737 (25.0%) | 208 011 (21.1%) |
| Number of sequences with source metadata present | Number of sequences with data linkages (optional) | ||||||
|---|---|---|---|---|---|---|---|
| Total number of sequences published | Isolate | Country | Collection date | Host | Isolation source (optional) | Biosample linkage present | SRA linkage present |
| 982 454 | 982 451 (99.9%) | 982 438 (99.9%) | 982 418 (99.9%) | 982 379 (99.9%) | 547 014 (55.7%) | 245 737 (25.0%) | 208 011 (21.1%) |
Submission statistics for sequences published by the NLM/NCBI SARS-CoV-2 GenBank processing pipeline, as of 21 October 2021
| Number of sequences with source metadata present | Number of sequences with data linkages (optional) | ||||||
|---|---|---|---|---|---|---|---|
| Total number of sequences published | Isolate | Country | Collection date | Host | Isolation source (optional) | Biosample linkage present | SRA linkage present |
| 982 454 | 982 451 (99.9%) | 982 438 (99.9%) | 982 418 (99.9%) | 982 379 (99.9%) | 547 014 (55.7%) | 245 737 (25.0%) | 208 011 (21.1%) |
| Number of sequences with source metadata present | Number of sequences with data linkages (optional) | ||||||
|---|---|---|---|---|---|---|---|
| Total number of sequences published | Isolate | Country | Collection date | Host | Isolation source (optional) | Biosample linkage present | SRA linkage present |
| 982 454 | 982 451 (99.9%) | 982 438 (99.9%) | 982 418 (99.9%) | 982 379 (99.9%) | 547 014 (55.7%) | 245 737 (25.0%) | 208 011 (21.1%) |
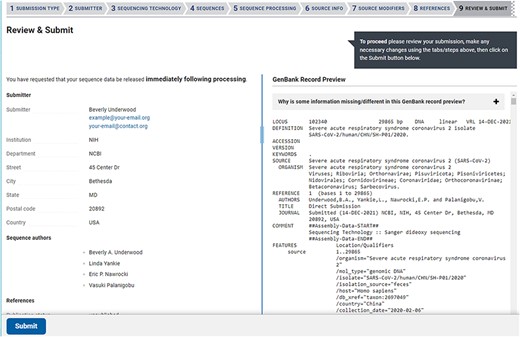
The Review & Submit page in a SARS-CoV-2 web submission. The left side of the page contains a summary of information, data files, and pre-submission sequence processing reports. The right side of the page displays a preview of the GenBank flatfiles prior to feature annotation and full submission processing. The preview is a familiar, human-readable format that allows submitters to confirm the author names, publications (if provided), contact information, sequencing technology and source information are correct before submitting.
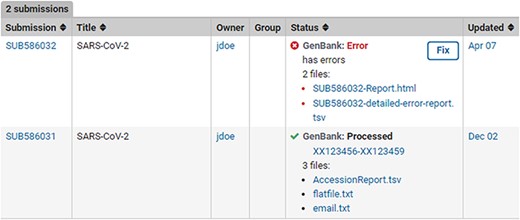
Post-submission processing reports. Example SUB586032 shows a (test) submission with errors with an error summary (SUB586032-Report.html) as well as a detailed VADR alert report (SUB586032-detailed-error-report.tsv). The error reports list the sequence_IDs with errors, instruction on how to correct the submission via the fix button or by contacting NLM/NCBI staff with more information. Example SUB586031 displays a successfully processed (test) submission with downloadable files containing GenBank Accessions, tab-delimited accession report with original sequence_IDs mapped to GenBank Accession numbers (AccessionReport.tsv), a copy of the fully processed annotated records (flatfile.txt) and a copy of the email sent upon successful processing (email.txt).
Performance and outcomes
At the time of this analysis (21 October 2021), the SARS-CoV-2 submission pipeline has processed and published to the database 982 454 SARS-CoV-2 sequences. To assess the quality and content of submissions processed through this pipeline, publicly available sequences processed by this software pipeline were downloaded and filtered from the NLM/NCBI Virus SARS-CoV-2 dashboard (https://www.ncbi.nlm.nih.gov/labs/virus/vssi/#/sars-cov-2). Submission statistics for data available at the time of this analysis is presented in Table 1.
Over 99% of the published sequences have relevant descriptive source metadata to give context to the sequence. Nearly all sequences (>99%) had values for the required source metadata fields, including country, collection date, host and isolate code. The records missing a value for a required field were evaluated to determine why data for a required field were missing. Two reasons were found to explain the missing values. In one case, the sequence was from a passaged source, so these fields were not applicable. However, metadata was present to accurately represent the laboratory passaging (e.g. MT576563). In other cases, host values were missing (75 sequence records). Records missing host information were generally from samples collected from the environment, which is more appropriately described in the isolation-source field (e.g. MT670008, from an air sample, not a host). The isolation-source field is not a required field for a SARS-CoV-2 submission. We observed around 55% of the submissions had a value for this non-required field. The data outcomes and usage patterns described above will be taken into consideration as we design future submission tools.
The submission pipeline optionally allows for submitters to provide the BioSample, BioProject, and SRA accessions. These accessions link the GenBank data with experimental data archived in other NLM/NCBI databases (11) giving database users direct access to the underlying data used to generate sequence assemblies and other experimental data from a given project or sample. Linking the assembled sequence to the underlying read data in SRA is an essential tool in tracking viral variation. At the time of this writing, over 20% of the sequences have these linkages (Table 1).
Processed data availability
Accessioned GenBank records are provided to the submitter for review and loaded to the public GenBank database for unrestricted access the day an error-free submission is made. Error-free submissions have a median processing time of 10 min, which means data are public approximately 10 min after a user submits the data for processing for web- and FTP-based submissions. Data published to GenBank are exchanged daily with INSDC partners ENA and DDBJ. Accessioned sequences subsequently become accessible via the NLM/NCBI SARS-CoV-2 resources page (https://www.ncbi.nlm.nih.gov/sars-cov-2/), NLM/NCBI Virus (https://www.ncbi.nlm.nih.gov/labs/virus/vssi/#/sars-cov-2), various searches (e.g. by accession number or organism), nr and betacoronavirus BLAST® databases (https://blast.ncbi.nlm.nih.gov/Blast.cgi) and NLM/NCBI Datasets (https://www.ncbi.nlm.nih.gov/datasets). The GenBank presentation of a SARS-CoV-2 record may include links to related resources, such as the BioSample, BioProject and/or SRA sequences associated with this record, if provided by the user. These linkages allow users to navigate between the raw reads and assembled genome, understand additional attributes about the sequenced sample and see other related sequences from the same initiative.
Conclusions
The SARS-CoV-2 submission pipeline allows submitters to quickly validate and publish partial and complete SARS-CoV-2 genome sequences to the public GenBank database through web-based forms or by FTP-based file deposition. Both methods provide automated feature annotation and sequence quality checks for the user. The web-based submission interface and links to other SARS-CoV-2 resources at NLM/NCBI may be found at https://www.ncbi.nlm.nih.gov/sars-cov-2/. The pipeline ensures the collection of a minimal set of specific source metadata to aid in maximizing the reusability of the data. By automating the submission, the pipeline also promotes the usage of consistent nomenclature and feature annotations and checks data for errors to allow for rapid release of quality SARS-CoV-2 data to the research community. NLM/NCBI has removed the burden of selecting source metadata and feature annotations for SARS-CoV-2 sequence submissions to streamline processing of these sequences. Similar automated processing strategies have been applied to other sequence data types for GenBank submissions, including ribosomal RNA, ribosomal RNA-internal transcribed spacer, influenza virus, dengue virus, norovirus and metazoan mitochondrial cytochrome oxidase subunit 1. We plan to continue to expand the processing capabilities to automate more sequence types in the future.
Funding
This work was supported by the National Center for Biotechnology Information of the National Library of Medicine (NLM), National Institutes of Health.
Conflict of interest
None declared.
Data availability
SARS-CoV-2 data resources are available at NLM/NCBI (https://www.ncbi.nlm.nih.gov/sars-cov-2/). SARS-CoV-2 sequence data may be submitted to GenBank via the web submission forms or FTP file deposition (https://submit.ncbi.nlm.nih.gov/sarscov2/genbank/). File creation, sequence screening and validation tools are available at the NLM/NCBI FTP site (https://ftp.ncbi.nih.gov/toolbox/ncbi_tools/converters/by_program/). The VADR annotation tool is provided on the NLM/NCBI GitHub repository (https://github.com/ncbi/vadr) and instructions for use on SARS-CoV-2 sequences are also on GitHub (https://github.com/ncbi/vadr/wiki/Coronavirus-annotation). SARS-CoV-2 submission processing errors are documented at NLM/NCBI (https://www.ncbi.nlm.nih.gov/genbank/sequencecheck/virus/ and https://www.ncbi.nlm.nih.gov/genbank/validation/). Accessioned sequences successfully processed through this pipeline are made available through NLM/NCBI Virus (https://www.ncbi.nlm.nih.gov/labs/virus/vssi/#/sars-cov-2), NLM/NCBI database searches (e.g. by accession number or organism in https://www.ncbi.nlm.nih.gov/nuccore/), nr and betacoronavirus BLAST® databases (https://blast.ncbi.nlm.nih.gov/Blast.cgi) and NLM/NCBI Datasets (https://www.ncbi.nlm.nih.gov/datasets).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}