





Abstract
Linking phenotypic with genotypic diversity has become a major requirement for basic and applied genome-centric biological research. To meet this need, a comprehensive database backend for efficiently storing, querying and analyzing large experimental data sets is necessary. Chado, a generic, modular, community-based database schema is widely used in the biological community to store information associated with genome sequence data. To meet the need to also accommodate large-scale phenotyping and genotyping projects, a new Chado module called Natural Diversity has been developed. The module strictly adheres to the Chado remit of being generic and ontology driven. The flexibility of the new module is demonstrated in its capacity to store any type of experiment that either uses or generates specimens or stock organisms. Experiments may be grouped or structured hierarchically, whereas any kind of biological entity can be stored as the observed unit, from a specimen to be used in genotyping or phenotyping experiments, to a group of species collected in the field that will undergo further lab analysis. We describe details of the Natural Diversity module, including the design approach, the relational schema and use cases implemented in several databases.
Introduction
In the last 20 years, high-throughput technology developments have revolutionized biology and transformed it into an information-based science. The first spur in data generation occurred in the early 1990's when large-scale sequencing became available through Sanger technology for relatively small-scale projects such as EST and BAC sequencing (1,2). In more recent years, the number of nucleic acids and protein sequences freely available at data repositories, such as GenBank, has grown exponentially as higher throughput and relatively inexpensive sequencing and genotyping platforms have enabled widespread generation of genomic data. Although Model-Organism Databases (MODs) were designed for storing genomic data and their derived annotations, they all face similar challenges in answering the needs of the developer and user community, including how to store the data efficiently, and how to adapt to newly emerging data types and represent them in a meaningful way so that biological questions can be answered.
At the core of the Generic Model Organism Database (GMOD) is a generic schema, named Chado, which was initially designed for storing Drosophila data at FlyBase, with the vision of creating a reusable and generic open source schema (3). Chado is ontology-driven and modular, and thus highly flexible. Chado's design principles enable the same schema to be used in projects with widely different metadata. Metadata can be modified or added as new data types become available. Its modular design allows developers to select those parts needed to manage their data, and to add new modules when advances in biology require new data types. Currently, the Chado schema consists of 18 modules, covering sequence, phenotype, genotype, ontologies, publications and phylogenies (http://gmod.org/wiki/Chado), with 23 genomic databases reporting that they use some or all of the Chado modules (http://gmod.org/wiki/GMOD_Users). As a GMOD component Chado is open source, and therefore any user can contribute to the schema and the underlying code (http://sourceforge.net/projects/gmod/), provided those contributions are consistent with the Chado generic design principle.
The initial development of Chado focused on genome sequence data, but as more complex data was generated, such as microarray and expression data, new modules were added to accommodate new data types. Chado has also proven useful for handling multiple closely related organisms. Clade Oriented Databases (CODs) using Chado include the Sol Genomics Network [SGN; http://solgenomics.net/ (4)]; Genome Database for Rosaceae [GDR; www.rosaceae.org (5)]; Citrus Genome Database (www.citrusgenomedb.org); Cool Season Food Legume Genome Database (www.gabcsfl.org); KnowPulse (http://knowpulse2.usask.ca/portal/) and the Genome Database for Vaccinium (www.vaccinium.org).
Unlike the exponential increase in sequencing data, phenotypic data has been growing at a much slower pace. Although count, structure and functional annotation for genes can be derived in silico using sequence similarity and other methods, analysis tools for correlating phenotype with genotype fall far behind those for sequence analysis (6,7).
A problem in phenotyping is the lack of genetic diversity in cultivated plants, which underwent heavy selection during domestication (8–11), causing a decrease in genotypic variation. As a result, cultivated plants may have as little as 5% of the natural diversity found in their wild counterparts. With such low allele pools, there is also a dramatic decrease in phenotypic variation. Another problem is related to difficulties in high-throughput phenotyping. For genotyping, one can choose from numerous available technologies according to desired quality and funds available. The end product is always a molecular sequence, and hence a uniform data type for which there are well understood and standard ways for processing and storage. In contrast, the data collection process for phenotyping is slow, expensive and subjective to the person collecting the data, generally with no set standards for terminology or descriptors to capture phenotype observations. Moreover, many phenotypes are subtle or even undetectable to the naked eye. Traits controlled by multiple quantitative trait loci may have dozens of underlying genes, each having a small additive affect (12). In addition, traits may be sensitive to environmental effects and may exhibit interactions with the genotype. Because of these and other challenges, phenotype data are notoriously difficult to record.
Although new technology, such as automated computerized facilities for growing plant germplasm (13) and software for taking multiple computerized measurements of a specimen (14), may facilitate high-throughput phenotyping, the challenge of capturing phenotypic diversity data remains complex, expensive and error-prone.
Despite these difficulties, breeding programs generate large volumes of phenotypic data, which poses a challenge for databases to efficiently store, query and analyze these data. In addition, such programs require genetic information to be integrated with phenotype data for progeny selection and crossing designs. Large-scale genotyping, mostly based on next-generation sequencing-based SNP markers, is now routinely performed on the same group of individual plants or animals for which phenotype assays were done. Large-scale phenotyping and genotyping experiments are currently practiced in various projects (15–18) as well as in applied breeding experiments. Shared among these projects is the challenge of determining how to best manage these data.
In maize, three monocot databases, Panzea (19), Gramene (20) and GrainGenes (21), jointly developed the Genomic Diversity and Phenotype Data Model (GDPDM) to capture molecular and phenotypic diversity data. The core schema of GDPDM consists of tables for germplasm, phenotype, genotype and environment that capture associations between phenotypes and genotypes. Although GDPDM performs well for its creators, the genericity of the schema is limited, and its design deviates enough from Chado's principles to make it difficult to adapt to other modules in Chado. As model organism and clade-oriented databases, which were already using Chado for storing and managing their data, increasingly faced the need for storing large-scale diversity data efficiently, several of them collaborated on developing a schema module for Chado capable of recording a wide variety of phenotyping and genotyping experiments in a way that maintains links to stocks and germplasms.
An initial version of a natural diversity module for Chado was developed at the National Evolutionary Synthesis Center (NESCent) in collaboration with W. Owen McMillan, who was a Center fellow at the time. McMillan is part of a community of researchers who use neotropical butterflies of the genus Heliconius as an emerging model system to study evolutionary genomics of Müllerian mimicry and adaptation (22,23). The data collected in this context resemble those of a clade-oriented database, and although not previously managed in GMOD, the need to connect the results of genetic and phenotypic diversity experiments and a growing body of molecular data strongly motivated modifying the data model as a future Chado Natural Diversity (ND) module. Later, several model organism (Medicago), clade-oriented (SGN, GDR, VectorBase) and plant-breeding (KnowPulse) databases formed a working group to collaboratively take up further development of the ND module. The goal of the working group was to mature the module to become an official part of Chado, fully consistent with its design principles and requirements, and as part of that expand its capacity for storing data from multiple experiments of specimens that were collected, treated and evaluated in various locations, environments and time points.
As a result of these revisions, the module now allows the storage of data from each experimental line that are scored for a large number of phenotypic traits, and genotyped with a set of genetic markers. In addition to storing data from experiments performed on existing lines, experiments that generate new lines and experimental samples, such as field collections, crosses and treatments, can be stored. In the remainder of the manuscript, the design of the ND schema and the use-cases from several of the databases participating in the working group are described.
Schema design approach
The development and maturation of the ND schema followed several guidelines imposed by best practices for Chado module design (flexibility, ontology-driven metadata, reuse of existing modules), and motivated by naming of schema elements that is intuitive yet remains unencumbered by potential name clashes with other modules. The following paragraphs present details on each of these.
Flexibility
As the ND module targets any type of experiment related to the collection of phenotypes and genotypes, the most important feature was to have a generic place for storing various assays. The ND schema was designed in a way that does not restrict the type of specimen that can be stored in the stock table and allows linking the specimen to any experiment via the nd_experiment_stock table. The ‘experiment’ name space does not necessarily refer to a complete biological experiment, but rather to an entity on the database level, sharing common attributes. For example, every time a phenotype or genotype is scored on a single stock, a new row is stored in the nd_experiment table. Likewise, an experiment can yield a new stock, thus the nd_experiment table is simply a placeholder allowing many-to-many relationships between stocks and experiments.
The nd_experiment table defines the type of experiment using a foreign key to the controlled vocabulary table (cvterm), eliminating the need for multiple tables for different types of experiments or assays, and also providing a link to the phenotype and genotype values.
The types of experiments tested during development of this module are:
phenotyping: any experiment involving scoring the subject for a trait value;
genotyping: any experiment yielding a value of the genetic makeup of the subject;
cross or mutation experiment: an assay involving crossing two subjects or mutation to generate new stocks; and
field collection: collection of data or specimens in the field for further analysis in the lab.
The generic design of the nd_experiment table, and optional many-to-many relationship between experiments and stocks, is flexible enough to allow other experiment types with the ND module.
Ontology-driven metadata
The ND module uses controlled-vocabulary (or ontology) terms for metadata rather than hard-coded column names whenever sensible. Metadata vocabulary terms are stored in the cvterm table, thus keeping domain entities unified and reusable. For example, if a stock is of type ‘plant accession’, this type is stored as a cvterm, which can then be used for other stocks of the same type. In the ND schema, the types of experiments and reagents are also modeled as links to the respective cvterms. The same approach is applied to all metadata properties of relational entities, for example dates or comments. Metadata properties are stored in property association tables, which are conventionally named by appending the suffix ‘prop’ to the name of the entity table for which it stores properties. For example, the table nd_experimentprop stores metadata properties for rows in nd_experiment.
Reusing tables from existing Chado modules
The ND modules reuse tables from the following modules, rather than redefining them: the stock module for stocks/specimens, the controlled vocabulary module for using ontology terms, and the genetic and phenotype modules for storing the relevant scores. The project table and the newly added project_relationship table are used to group similar experiments. The contact and publication modules can be used to record contact and reference information for experiments.
ND prefix for the tables in the new module
The ND module introduces ‘nd_’ as a prefix for table names to allow intuitive naming of tables (and related relational entities such as primary key sequences) whereas avoiding easily conceivable collisions with tables in other modules. For example, the central table of ND is ‘experiment’, which as a term is common to many areas of experimental biology but has sufficiently different semantics depending on the biological application that using one and the same table across all Chado modules that refer to an experiment would be prone to confusion. Trying to choose a distinct name can easily suggest inaccurate semantics although not necessarily avoiding the name collision problem; for example in earlier versions the nd_experiment table was named ‘assay’, which is less accurate yet still conflicts with the Chado MAGE module for microarray data. As the Chado database schema expands to more kinds of biological data, prefixing the names of tables and primary keys may become a new recommended practice for Chado modules.
Schema description
The ND module consists of tables, which are used to store data from various experiments performed on biological entities and also track the relationships of experiments with data stored in other modules. Other key modules that are integral to the ND module include the Stock, Phenotype and Genetic modules (Figure 1). For current status of the ND schema see the GMOD wiki module page (http://gmod.org/wiki/Chado_Natural_Diversity_Module). Latest version of the Chado schema can also be found on the GMOD website (http://gmod.org/wiki/Downloads).
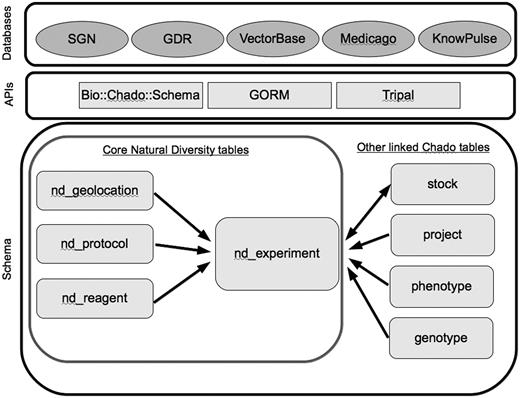
Core tables in the Natural Diversity module, and directly linked tables from other Chado modules. Databases are the five member organizations of the Natural Diversity development working group, and the APIs are some of the solutions currently used for interacting with the Chado schema.
Natural diversity module
Experiments and their relationship with other data
The nd_experiment table is central to the ND module. The notion of experiment in this module is conceptual, and may not reflect a whole biological experiment. Each nd_experiment has a type (common types are phenotyping, genotyping, field collections, crossing, mutation and propagation), and a location. As a generic guideline whenever a person collects data at a certain date and location, a new nd_experiment row is stored. The existing Chado project table is used for storing the top-level experimental design, and nd_experiment_project table for grouping nd_experiments and associating those with the project (Figure 2).
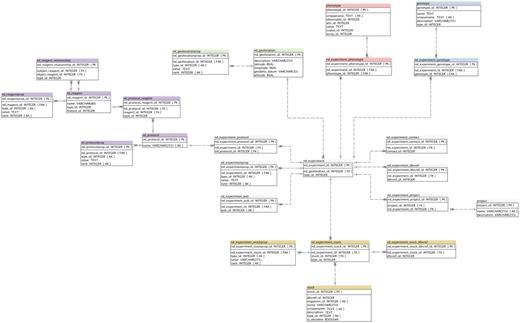
Detailed Schema diagram of the Natural Diversity Module, and interactions with other major Chado modules. All Natural Diversity tables are denoted with ‘nd_’ prefix.
Another important feature of experiments in ND is the ability to maintain many-to-many relationships between each nd_experiment and stocks using the nd_experiment_stock linking table.
One stock entry can be linked to multiple experiments through the nd_experiment_stock table. For phenotyping and genotyping experiments, many times these will be linked with a specific protocol. In such cases it is recommended that each row in the nd_experiment be linked to a single row of stock table and to a single row of genotype or phenotype table. Exceptions to this would include where a row of the nd_experiment is linked to multiple rows of the genotype table to store the genotype of a heterozygote. In this case, the genotype of each allele of the heterozygote is stored in a distinct row of the genotype table. However, users can choose to store the genotype of a heterozygote in one row of the genotype table. Multiple phenotypes can be linked to a single nd_experiment row when there is no need to store a specific protocol, for example when measuring lengths of several plant parts.
A row of nd_experiment of type ‘genotyping’ or ‘phenotyping’ represents the particular experiment performed on a specific sample of a biological entity. The associated metadata of an experiment such as the experimenter, date and comments can be stored in the nd_experimentprop table. When a stock was subjected to any biotic or abiotic treatment prior to phenotypic evaluation, it can be linked to an experiment of type ‘sample_treatment’ and the metadata and the protocols of the sample treatment experiment can be stored in nd_experimentprop, protocol and protocolprop (see section 1.2). Since one row of the experiment of type ‘genotyping’ or ‘phenotyping’ describes an assay that uses a single stock_id to give rise to a single genotype or phenotype, multiple nd_experiments that relate to each other can be linked to the same record in the ‘project’ table. A project can consist of several smaller sub-projects and the relationships among projects can be stored in the project_relationship table. The nd_geolocation table stores the details of the geolocation where the experiments were carried out, such as the geolocation of a field where plants were grown.
Protocols, Reagents and their relationship with other data
The nd_protocol table stores any procedure performed as part of an experiment. Specific reagents used in a protocol can be stored in the nd_reagent table. Multiple rows of the nd_experiment table can be linked to a single row of the nd_protocol table, and multiple rows of the nd_protocol table can be linked to a single row of the nd_reagent table (Figure 2).
Stock module
The original stock module was designed to store information about stock collections in a laboratory. This original concept of ‘stock’ has been expanded to accommodate conceptual entities that a stock belongs to or entities that were derived from a stock for specific experiments. Hence, the stock table can store hierarchical entities of population, strain/line/accession, individual, clone and sample, with relationships between stocks defined in the stock_relationship table. For example, a stock could be a population maintained in a bottle in the lab composed of a mixture of multiple genotypes, or could be an inbred line of a plant whose genotype is fixed. In a phenotyping experiment of plants in field plots, with 10 plants for each genotype, one would want to store each plant separately, since the phenotype is recorded on the individual plants. This method allows keeping track of the seeds of the progeny, collected from each plant for future experiments based on analysis results. In this case, each plant may be stored as a stock, with the parent line indicated in stock_relationship (e.g. object_id is the parent accession, subject_id is the plant in the field plot, with type_id of ‘plot_of’).
Since a population can be defined as a group of entities, a population entity can be composed of multiple species such as a group of insects collected in a field. To accommodate this concept, the ‘not null’ constraint for organism_id of the stock table has been dropped. Any metadata of a stock can be stored in ‘stockprop’ table (http://gmod.org/wiki/Chado_Stock_Module).
Phenotype module
The phenotype table is linked to the ND module via the nd_experiment_phenotype table. The descriptors of phenotypes can be stored using observable_id and/or attr_id of the phenotype table, and the specific phenotypic value of a biological entity from a specific experiment is stored in phenotype.value or phenotype.cvalue_id. The detail of the descriptors (observable_id and attr_id) can be stored in the cvterm table (http://gmod.org/wiki/Chado_Phenotype_Module).
The phenotype module was designed and used by FlyBase. Although the ND working group designed the ND module, a few basic issues concerning the phenotype table were raised. Currently, the phenotype table holds both the phenotype value, and potentially a post-composed cvterm [e.g. observable_id = ‘fruit_id’ from the Plant Ontology (PO), attr_id = ‘color’ from the Phenotype And Trait Ontology (PATO)]. This design does not allow reusing post-composed terms, and does not separate the phenotype value from the descriptor. Since this problem does not have one agreed upon solution, and it is beyond the scope of the ND module, a new revision of the Chado phenotype module is required. Since revisiting the phenotype module may cause backward compatibility issues for existing users, any changes require careful documentation and migration scripts for updating the schema. Moreover, schema changes should deprecate, rather than delete, existing tables or columns.
Genetic module
The genotype table is linked to the ND module via the nd_experiment_genotype table. For large-scale genotyping data using molecular markers, the allelic variation can be stored in genotype.description and the markers that are stored in the feature table can be linked to the genotype table via the feature_genotype table (see the Chado genetic module http://gmod.org/wiki/Chado_Genetic_Module).
Use cases (example data representations)
The use cases given below are some examples of how various biological data can be stored in the ND module and other related Chado modules. Although the examples below of ND ‘best practices’ are the results of numerous discussions, meetings and different database and user requirements, the system provides flexibility, allowing the inclusion of other data storage approaches.
ND usage relies heavily on the stock module. In each of the following use cases, samples or accessions are stored in the stock module, and phenotype/genotype experimental results are stored in the ND module. The unique two-way linking table, nd_experiment_stock, allows storing experiments of existing stocks, as well as generating new stocks from various experiments, such as field collections.
The various queries below simply illustrate how the data are stored but do not necessarily represent the best way to query the data. Materialized views can be created for faster query performance.
Plant breeding data (Genome Database for Rosaceae)
Genome Database for Rosaceae (GDR; http://www.rosaceae.org/) contains genetic and genomic data of the Rosaceae family (almond, apple, apricot, blackberry, cherry, peach, pear, plum, raspberry, rose and strawberry). Currently, GDR also contains private breeding data from individual breeders and public breeding data from the RosBREED project (15). In fruit tree breeding programs, progeny of tree fruit breeding crosses are used for genotype and phenotype evaluations. The individuals of progeny are clonally propagated and planted in multiple locations so that breeders can obtain adequate quantity for further evaluations. Phenotype evaluations are done multiple times on the same clones. Samples can be collected from the same clone multiple times and the subsets of samples that are collected at the same time can be treated in different conditions before phenotypic evaluation. Genotyping can also be done multiple times on the same variety.
Pedigree and cross data
The pedigree data of varieties are stored using the stock_relationship table. The example query below shows the pedigree information of the variety ‘T1119’.
SELECT s1.uniquename AS subject, c.name AS relationship, s2.uniquename AS object
FROM stock s1
JOIN stock_relationship sr ON s1.stock_id = sr.subject_id
JOIN stock s2 ON sr.object_id = s2.stock_id
JOIN cvterm c ON sr.type_id = c.cvterm_id
WHERE s2.uniquename = T1119
OR s1.uniquename = T1119
| Subject | Relationship | Object |
|---|---|---|
| Splendour | is_a_maternal_parent_of | T1119 |
| Gala | is_a_paternal_parent_of | T1119 |
| T1119 | is_a_maternal_parent_of | C1111 |
| T1119 | is_a_paternal_parent_of | C1112 |
| T1119 | is_a_maternal_parent_of | C1113 |
| T1119 | Is_a_mutation_parent_of | C1114 |
| Subject | Relationship | Object |
|---|---|---|
| Splendour | is_a_maternal_parent_of | T1119 |
| Gala | is_a_paternal_parent_of | T1119 |
| T1119 | is_a_maternal_parent_of | C1111 |
| T1119 | is_a_paternal_parent_of | C1112 |
| T1119 | is_a_maternal_parent_of | C1113 |
| T1119 | Is_a_mutation_parent_of | C1114 |
Crossing experiments
Crosses between accessions are stored as an nd_experiment with type of ‘cross experiment’, and linked to the parent accessions via the nd_experiment_stock. Derived progeny can then be stored in the stock and stock_relationship tables. This practice allows keeping track of pedigrees and progeny accessions used in subsequent experiments.
Phenotype evaluation
In fruit tree breeding, a variety is usually propagated to produce multiple clones and the phenotype of each clone is evaluated multiple times. Each sample that gives rise to one phenotypic value is associated with one nd_experiment. The sample can be stored in the stock table and the relationships among varieties, clones and samples can be stored in the stock_relationship table. The trait descriptors developed by individual breeders are stored in the cvterm table, and linked from phenotype.attr_id. Currently, Solanaceae breeders’ ontology exists, and Rosaceae breeders also developed standard trait descriptors as part of RosBreed project, which are also stored in the cvterm table. Phenotyping data from individual breeders can be stored using standard descriptors as well as their individual descriptors, so that breeders can choose to view the data either way. When breeders want to compare their results with those from other breeders, they can choose to view the data in standard descriptors. Metadata of the samples, such as harvest dates, are stored as properties of the stock (stockprop table).
An example query below shows all the phenotypes that are associated with a sample. If we want to query all the phenotype data for a specific variety, we can query all the phenotypic values of all the samples of a specific variety.
SELECT s.uniquename AS sample, c1.name AS phenotype, p.value AS value
FROM stock s
JOIN nd_experiment_stock es ON s.stock_id = es.stock_id
JOIN nd_experiment_phenotype ep
ON es.nd_experiment_id = ep.nd_experiment_id
JOIN phenotype p ON ep.phenotype_id = p.phenotype_id
JOIN cvterm c1 ON p.attr_id = c1.cvterm_id
WHERE s.uniquename = T1120
| Sample | Phenotype | Value |
|---|---|---|
| T1120 | SWEET | 3 |
| T1120 | OVERALL | 1 |
| T1120 | GRDCOL | 1.5 |
| T1120 | CRISP | 2 |
| T1120 | JUIC | 3 |
| T1120 | HUE | 5.50 |
| Sample | Phenotype | Value |
|---|---|---|
| T1120 | SWEET | 3 |
| T1120 | OVERALL | 1 |
| T1120 | GRDCOL | 1.5 |
| T1120 | CRISP | 2 |
| T1120 | JUIC | 3 |
| T1120 | HUE | 5.50 |
QTL and association mapping data (SGN)
The Sol Genomics Network (SGN; http://solgenomics.net) database hosts an extensive phenotype and genotype database of Solanaceae plant accessions (4,24). Much of these data originate from breeding experiments of important crops of the family, mainly tomato and potato. One of the major goals is to collect the phenotypes and genotypes in standard formats for linking phenotypic variation with genetic markers, and eventually identify the underlying genotypes.
A problem with acquiring breeding data from multiple resources is how to represent phenotypic scores of germplasm planted and collected by different people at different locations, dates and environments. The Chado ND module can address these issues.
Phenotyping field experiments
Usually plant-breeding data is collected for single plants, situated in a field or greenhouse, with a predetermined experimental design of planting pattern to enable statistical analysis whereas eliminating potential interfering factors, such as location in the field. Each assayed plant is stored in the stock table, and stock_relationship holds the link to the parent accessions. Then each field experiment is stored in the nd_project table for grouping all subsequent phenotyping and genotyping experiments on each individual plant in the field plot.
The table nd_experiment is used as a placeholder for one phenotyping event or assay performed on one plant stock. Usually if several phenotypic scores are measured for one plant by one person (or computer software) on the same date and location, then one row is stored in nd_experiment and nd_experiment_stock, and each phenotype is recorded separately in the phenotype table. All the phenotypes are linked to the experiment via the nd_experiment_phenotype table.
Phenotypes may be quantitative or qualitative. The Chado phenotype module does not distinguish between the two modes, and the phenotypic value is stored in the phenotype.value field as a character, whether the value is numeric continuous, ordinal or a string descriptor. The phenotype is stored as Entity-Quality (EQ), and value whenever applicable. Entity is the observable, and usually a Solanaceae Phenotype Ontology term (SP) is used (http://solgenomics.net/search/direct_search.pl?search = trait), which is a breeder-focused vocabulary developed according to user requirements (e.g. Plant habit index SP:0000128), essentially created as pre-composed terms taken from the Plant Ontology and PATO. The quality is the attribute, stored as a Phenotype and Trait Ontology term (PATO), or as a Solanaceae Phenotype term, which is mapped to a PATO term (e.g. SP:0000196 ‘spreading’ is mapped to PATO:0001855 ‘horizontal’). This SP to PATO mapping allows SGN to give breeders direct access to trait descriptors, eliminating the need to use multiple vocabularies, as well as using very granular and Solanaceae specific trait values (e.g. SP:0000332 ‘spreading habit 30-15 degrees’).
Although statistical analysis of quantitative phenotypes is required for QTL detection and mapping, there is a need to filter the phenotypes linked to nd_experiments, and subsequently to stocks, such that only quantitative numeric phenotypic values are included.
A sample query and results for finding the average phenotypes by quality is given below for retrieving the traits of the processing tomato accession ‘Saucy’. Since phenotypes are not scored directly on the stock for the accession, but rather on individual plants as observational units (‘subjects’) the query finds all the phenotypes of the accession's subjects. Further filtering is possible by project, especially if different plants were scored at different locations of different environmental effects.
SELECT attr.name AS attribute, avg(cast (phenotype.value as real))
FROM stock JOIN stock_relationship ON stock_id = subject_id
JOIN nd_experiment_stock USING (stock_id)
JOIN nd_experiment USING (nd_experiment_id)
JOIN nd_experiment_phenotype USING (nd_experiment_id)
JOIN phenotype USING (phenotype_id)
JOIN cvterm AS attr ON attr_id = attr.cvterm_id
WHERE object_id = (SELECT stock_id FROM stock WHERE name = 'Saucy')
GROUP BY attribute;
| Attribute | Average |
|---|---|
| Horizontal asymmetry ovoid | 3.66 |
| Heart shape | 0.55 |
| Proximal fruit end indentation | 0.03 |
| Cross section average chroma | 36.89 |
| Cross section average 'b' value | 27.98 |
| Cross section average L value | 37.49 |
| Proximal angle macro 20% | 92.88 |
| Attribute | Average |
|---|---|
| Horizontal asymmetry ovoid | 3.66 |
| Heart shape | 0.55 |
| Proximal fruit end indentation | 0.03 |
| Cross section average chroma | 36.89 |
| Cross section average 'b' value | 27.98 |
| Cross section average L value | 37.49 |
| Proximal angle macro 20% | 92.88 |
Genotyping data
The individual plant samples that are phenotyped are also often genotyped. The genotype scores for each marker are stored in the nd_experiment table with a type_id of 'genotyping experiment', which are linked to the Chado genetic module via nd_experiment_genotype.
In QTL studies, phenotypes are correlated with differences in genotypes (genetic markers showing polymorphisms). The ND schema design allows simple querying of the data required for running statistical methods for finding traits which co-occur with a marker or a location on the genetic map. Currently, a module exists that performs QTL analysis for F2 and Back-cross bi-parental plant populations with continuous phenotypic variation (25).
For association analysis of populations of unknown structure, similar phenotype and genotype experimental data can be used. Although more elaborated statistical methods are required for association-mapping, all the necessary data for this analysis can be easily queried in a manner similar to the QTL tool. Existing tools, such as TASSEL (26), can then be applied to the data, and new statistical tools, for example, based on R (http://www.r-project.org/), will be implemented.
Phenotypic assay of wild-caught samples (VectorBase)
A large proportion of the data in VectorBase (http://www.vectorbase.org/) (27) concerns insecticide resistance. The process of assessment is generally three-fold. Mosquitoes are collected from the field, their species is identified and then they are subjected to the insecticide resistance assay. The ND schema is well suited to capturing all of these aspects.
Each of these actions will be recorded as a separate entry in the nd_experiment table, such that a typical sample will be connected to one field collection assay, one species identification, and one phenotypic assay.
Field collection metadata
In addition to the field collection information, it is important to record the catch method and other metadata since it can significantly affect the types of samples found or have implications for the phenotype itself (e.g. a collection performed inside a dwelling strongly suggests an anthropophilic mosquito). This is stored in the nd_protocol / nd_protocolprop tables using terms from one of the vector biology-specific ontologies such as MIRO (28) (e.g. MIRO:30000009 – ‘indoor light trap catch’).
The site of the field collection is stored in the nd_geolocation and nd_geolocationprop tables. The level of detail is left up to the individual submitters, but the nd_geolocation table expects a latitude and longitude, along with the geodetic datum (e.g. ‘WGS 84’) and a gazetteer ID is a required field in VectoBase to specify location names.
Species Identification
Determining which of the morphologically identical species have been captured is most often achieved by comparing species-specific ribosomal DNA sequence, and would be recorded in the nd_protocol/nd_protocolprop table in a similar manner to the field collection method (e.g. MIRO:30000040 – ‘PCR-based species identification’). Species can therefore be recorded in two different places, either in the nd_experiment_genotype/genotype tables, or directly in the stock/organism tables.
In cases where a species identification assay has been provided the genotype tables should be used to record the direct result of this assay and an NCBI taxon ID if known. Where a stock species is known – regardless of whether we have assay data for this or not - this should be recorded in the stock table using the relevant NCBI taxon ID. It is worth noting that, although the organism field can be left null, it is informative – that the Spp. has not been determined unambiguously.
Phenotypic assay
Wild-caught mosquitoes are assayed for resistance to insecticides using a number of different methods. Many of these are standardised by the WHO (World Health Organization) and as such would be linked to the same records in the protocol table. Factors that change from assay to assay, such as the precise number of mosquitoes used or any varying insecticide concentrations would be recorded in the nd_experimentprop table.
Though these assays may use different metrics, they are all an attempt to measure the same phenotype: the relative level of tolerance to the insecticide. As a result VectorBase stores this information both as numerical measurements and also as linked Entity-Quality (E-Q) values using dedicated vector biology ontologies [E] and PATO [Q]. In this way two methods may use different metrics (e.g. MIRO:20000013 Time Response Test may be measured in seconds, and MIRO:20000076 Dose Response Test may be measured in % mortality) yet the resultant phenotype for both could be the same (e.g MIRO:00000003/PATO:0001650—metabolic resistance/increased resistance).
Large-scale phenotyping and genotyping data (Medicago ecological genomics data)
The model legume Medicago truncatula is an emerging system for ecological genomics and association mapping, since the genomes of at least 400 inbred lines will be re-sequenced and polymorphism data will be linked with phenotypic variation. Data are currently available for 40 inbred genotypes that have been grown under a range of environmental conditions and phenotyped for the same suite of traits that have been formalized into a custom ontology. Treatments include different field sites, manipulated NaCl concentrations in the greenhouse, and varying NaCl environments experienced by the maternal plant of an individual seed.
The project table is used for grouping a set of experiments, and each of those set consist of several assays performed on one individual. The information about each individual is stored in the stock table with a type_id of ‘inbred line’. Properties of the individual, such as ‘plant id’, ‘location in holder’, and ‘soil replaced date’, are stored in the stockprop table. The nd_experiment is linked to the stock via the nd_experiment_stock table. Genotyping and phenotyping assays are stored in nd_experiments with links to the genotype and phenotype tables respectively.
The type of treatment applied to the individual stock is stored in the nd_protocol table using the type_id field (e.g. ‘NaCl treatment’). The reagent ‘NaCl’ is stored in the nd_reagent table, and the treatment is linked to nd_experiment with a type_id of ‘treatment’ via the nd_experiment_protocol table.
Phenotype descriptions are stored in the Phenotype table. An example of a phenotype description is ‘stem diameter at harvest (mm)’. This description is decomposed into an entity-quality term ‘stem diameter’, a temporal modifier ‘at harvest’, and unit ‘mm’. The EQ term is defined in our Diversity Experiment Ontology (DEO). The term id is stored in the observable_id field, and the unit ‘mm’ is defined in the Unit Ontology (UO). The modifier ‘at harvest’ is composed of two subterms, a relation ‘at’ (or its synonym ‘exists_at’) and the temporal quality ‘harvest’. Both of these terms are defined in the DEO.
Each phenotyping assay (nd_experiment) is linked to a phenotype via the nd_experiment_phenotype table. Statements linking the environment, genotype, and phenotype, are stored in the phenstatement table. For example, a statement of this type would be “The mean of the phenotype flower number in genotype TN7.4 given an environment of NaCl treatment of 100 millimolar is 10”. A row with uniquename of ‘high salt’ is stored in the environment table. A row with uniquename set to the project name and type_id of ‘experimental result’ is stored in the pub table.
Genotype Evaluation (KnowPulse; http://knowpulse2.usask.ca/portal/)
Genotypic assays are usually performed with DNA from a given individual. Often DNA is extracted mutliple times from a given individual and then multiple genotypic assays may be performed on each DNA or cDNA sample. Both the individual and each DNA sample from that individual are stored in the stock table and the samples are related back to the individual from which they were taken using the stock_relationship table.
The basic goal of a genotypic assay is to identify the genotype of a given individual at a given genetic locus where a genetic locus is defined as a position in the genome of the organism. Genetic loci are stored in the feature table (See Chado Sequence module). A list of genetic loci available for screening for a given individual (Sheyenne) can be obtained by the following example query.
SELECT f.name, f.uniquename, fl.fmin as position_start, fl.fmax as position_end, srcf.name as feature_located_on
FROM feature f
JOIN cvterm t ON t.cvterm_id = f.type_id
JOIN featureloc fl ON fl.feature_id = f.feature_id
JOIN feature srcf ON srcf.feature_id = fl.srcfeature_id
JOIN stock s ON s.organism_id = f.organism_id
WHERE s.name = 'Sheyenne'
AND t.name = ‘genetic_marker’;
The genotype observed for a given loci is stored in the genotype table (See Chado Genetic module) with the description of the genotype observed, whether that be a sequence, presence/absence or some other standard description. The locus for which the genotype was identified is linked to the genotype through the feature_genotype linking table. An example query listing all the observed genotypes for a given locus is shown below.
SELECT g.name, g.uniquename, g.description as genotype
FROM genotype g
JOIN feature_genotype fg ON fg.genotype_id = g.genotype_id
JOIN feature f ON f.feature_id = fg.feature_id
WHERE f.uniquename = 'SUKHA-1';
The sample stock is linked to the observed genotype through the nd_experiment table since a genotypic assay was performed in order to determine what the observed genotype for a given sample is. Thus, as with phenoypic data, a single experiment in the nd_experiment table identifies the genotype of a single genetic locus in a single sample. The protocols of the genotypic assay can be stored in nd_experiment and other associated tables (See use cases of Medicago ecological genomics data). The sample is linked to the experiment through the nd_experiment_stock linking table and the observed genotype is linked to the experiment through the nd_experiment_genotype linking table. An example query is shown below which lists all of the stocks that have been assayed at a given locus including the genotype observed and the experiment that identified the genotype.
SELECT geo.description as location, expt.name as experiment_type, s.name as stock_name, s.uniquename as stock_uniquename, g.description as genotype
FROM nd_experiment exp
JOIN nd_geolocation geo ON geo.nd_geolocation_id = exp.nd_geolocation_id
JOIN cvterm expt ON expt.cvterm_id = exp.type_id
JOIN nd_experiment_stock exps ON exps.nd_experiment_id = exp.nd_experiment_id
JOIN stock s ON s.stock_id = exps.stock_id
JOIN nd_experiment_genotype expg ON expg.nd_experiment_id = exp.nd_experiment_id
JOIN genotype g ON g.genotype_id = expg.genotype_id
JOIN feature_genotype fg ON fg.genotype_id = g.genotype_id
JOIN feature f ON f.feature_id = fg.feature_id
WHERE f.name = 'SUKHA-1';
Application Programming Interfaces
The ND module provides a generic schema for storing experimental results. Most resources using this schema will require front-end applications with graphical interfaces for their users and possibly for data curation purposes. Application Programming Interface (API) development is independent from the schema, as long as the software knows how to connect, query, and fetch results from the database back-end. The following list, not meant to be exhaustive, describes some examples of the ND schema APIs available for different programming languages.
- (1)
Bio::Chado::Schema (http://gmod.org/wiki/Bio::Chado::Schema)
Bio::Chado::Schema is a GMOD project that provides a standard object-relational mapping (ORM) layer in Perl for the Chado schema. It is implemented using DBIx::Class (http://www.dbix-class.org/) and provides support for all Chado modules, including the ND module.
- (2)
Grails’ Object Relational Mapping (GORM) and Hibernate
Hibernate is a Java ORM. The Hibernate Tools can be used in Eclipse or Ant to reverse engineer the Chado schema into Hibernate domain classes. GORM is part of the Groovy/Grails web framework. It can use the Hibernate classes to provide simple findByAttribute queries, criteria queries involving more than two attributes, and queries via the Hibernate Query Language (HQL).
- (3)
Drupal and Tripal (http://gmod.org/wiki/Tripal)
Drupal is a content management system that provides various practical benefits as a front-end for Chado. Tripal (29) is a GMOD project that serves as a web front end based on a collection of Drupal modules. The current version supports various visualizations for various Chado modules and further development is underway to support ND module.
Discussion
The ND Module was developed to store large-scale phenotyping, genotyping and breeding data. Even though the existing Chado modules allow storing the genotype and phenotype of stocks, it cannot accommodate multiple genotypic and phenotypic values of stocks and experimental metadata that are generated by large-scale experiments. To meet these needs, the Chado ND Module Working Group was formed, which consists of database developers interested in using Chado to store their large-scale phenotyping and genotyping data, to collaboratively develop a new module. Since the working group consists of developers dealing with diverse organisms, projects and experimental designs, the module was naturally conceived with flexibility in mind.
The starting point was a ND module created in 2007 for managing genetic and phenotypic diversity data collected for Heliconius butterflies, an emerging model system for evolutionary genomics of mimicry and adaptation (22,23). Subsequent work focused on maturing the ND module into an officially accepted Chado module fully consistent with Chado's design principles and requirements (3). This included changes so that it can accommodate a wider variety of genotyping and phenotyping experiments as well as new types of stock with their associated phenotypes and genotypes. A key concept in the module's current design is to use the nd_experiment table for any kind of experiment or assay that uses or produces stocks. A single row of nd_experiment is created to link one stock to one distinct phenotypic or genotypic value, stored in the phenotype or genotype table. Another key concept is the capacity to store biological entities of any hierarchical level in the stock table, and to store groups of experiments of any hierarchical level in the project table, just as features in the genome of any hierarchical level can be stored in the feature table. This design gives flexibility to accommodate phenotyping and genotyping data from diverse types of projects.
One of the main purposes developing open-source generic schemata for biological data is to promote sharing of software that work with the schema, as well as to provide generic schema to develop new databases (30,31). However, due to the flexibility of the schema, there are many different ways to store large-scale phenotyping and genotyping data, which may present an obstacle to efficient sharing and reusing of software. To provide future database developers with some guideline and examples, this article includes various use cases adopted by diverse databases. An important future effort to promote the standardization of the usage of the schema includes developing ontology to store various types of stock and their relationships.
Although the level of normalization of the ND schema allows storing data in the magnitude of millions of entries, database performance is still an issue yet to be addressed, especially with very large data sets, such as SNP genotyping. Other database management systems for genotype/phenotype data, such as the GDPDM schema (19), should have similar performance issues, thus MOD users and developers will have to set methodologies for efficiently storing SNP and next-generation sequencing data (32), since all raw data should probably not be stored as-is in the database.
The ND module was developed with plant breeding data (Solanaceae, Rosaceae and legume species) and large-scale natural diversity data (mosquitoes) in mind. These types of data are expected to grow rapidly due to the advance of high-throughput technology and the increasing effort toward marker-assisted breeding. The participating databases include GDR (5), SGN (4) and KnowPulse (http://knowpulse2.usask.ca/portal/). The collaboration was done mainly through conference calls and email correspondences. The development process was very efficient and productive; presenting this type of collaboration as a good model for further development of open-source bioinformatics tools.
Software development and database management are the responsibility of each participating database. Although there is no one standard API for the ND module (and Chado in general), or even a standard programming language or Database Management System (DBMS), there are several software packages and modules that were written to interact with Chado, yet these are distributed separately. Some examples are Bio::Chado::Schema and Bio::DB::Das::Chado, both available from CPAN (http://search/cpan.org) and Modware, available at Sourceforge (http://sourceforge.net/projects/gmod-ware/). The ND module is an integrated part of GMOD and the Chado schema, and can be downloaded and updated from SVN (http://gmod.org/wiki/Chado#Chado_From_SVN), or downloaded as part of the periodically updated Chado schema stable release (http://gmod.org/wiki/Downloads). Since Chado is a part of the GMOD open-source project (http://gmod.org), it relies heavily on the user and developer communities to test, use, fix and contribute to both schema and code. See the GMOD web site for ways of obtaining help, or join the Natural Diversity mailing list (https://lists.sourceforge.net/lists/listinfo/gmod-phendiver).
ND module is expected to be revised and improved as more users may display other needs for data storage and analysis. As with any other open-source project, the success of this collaboration relies on future maintenance and software development by GMOD users. The participating groups in this project are currently writing loading scripts, APIs and documentation for ND module ‘best practices’. Developing open-source code, which interacts with the schema (e.g. the code behind SGN is freely available from github https://github.com/solgenomics; 4), will make this project more sustainable in the long run, as more users show interest in a platform for phenotype to genotype analyses.
Funding
This work was supported by the informal collaboration between various institutions. Individual who have contributed to the development of Natural Diversity Module received from various sources including (USDA) SolCAP grant (Project Number: 5366-21000-026-09) (SGN); the USDA NIFA SCRI (grant # 2010-2010-03255) (GDR and SGN); the Agriculture and Agri-Food Canada (AAFC) Grant: ‘Purenet’ under the Agricultural Bioproducts Innovation Program (ABIP) (2009–2011) (KnowPulse); NSF DBI (grant 0820846) (Medicago ecological data); NESCent (NSF EF-0905606 and EF-0423641); and NIAID contract to VectorBase. Schema development was performed as part of the GMOD project. GMOD is supported by a specific cooperative agreement from the USDA Agricultural Research Service, and by NIH grants co-funded from the National Human Genome Research Institute and the National Institute of General Medical Sciences. Funding for open access charge: USDA NIFA SCRI (grant # 2010-2010-03255).
Conflict of interest. None declared.
Acknowledgements
We thank W. Owen McMillan (Smithsonian Tropical Research Institute), Chris Jiggins (University of Cambridge), Alexie Papanicolaou (CSIRO) and Nassib Nassar (Renaissance Computing Institute) for their invaluable contributions to early versions of the ND schema module.
Please note: A more legible, high resolution version of Figure 2 has replaced the original published version. The content has not been changed.
References
Author notes
†These authors contributed equally to work.
‡Present address: Seth Redmond, Pasteur Institute, 28 Rue Du Docteur Roux, Paris, 75015, France.
¶Present address: Dave Clements, Department of Biology, Emory University, Atlanta, GA 30322, USA.

{kind=link}
{kind=link}