





Abstract
Understanding how cellular metabolism works and is regulated requires that the underlying biochemical pathways be adequately represented and integrated with large metabolomic data sets to establish a robust network model. Genetically engineering energy crops to be less recalcitrant to saccharification requires detailed knowledge of plant polysaccharide structures and a thorough understanding of the metabolic pathways involved in forming and regulating cell-wall synthesis. Nucleotide-sugars are building blocks for synthesis of cell wall polysaccharides. The biosynthesis of nucleotide-sugars is catalyzed by a multitude of enzymes that reside in different subcellular organelles, and precise representation of these pathways requires accurate capture of this biological compartmentalization. The lack of simple localization cues in genomic sequence data and annotations however leads to missing compartmentalization information for eukaryotes in automatically generated databases, such as the Pathway-Genome Databases (PGDBs) of the SRI Pathway Tools software that drives much biochemical knowledge representation on the internet. In this report, we provide an informal mechanism using the existing Pathway Tools framework to integrate protein and metabolite sub-cellular localization data with the existing representation of the nucleotide-sugar metabolic pathways in a prototype PGDB for Populus trichocarpa. The enhanced pathway representations have been successfully used to map SNP abundance data to individual nucleotide-sugar biosynthetic genes in the PGDB. The manually curated pathway representations are more conducive to the construction of a computational platform that will allow the simulation of natural and engineered nucleotide-sugar precursor fluxes into specific recalcitrant polysaccharide(s).
Database URL: The curated Populus PGDB is available in the BESC public portal at http://cricket.ornl.gov/cgi-bin/beocyc_home.cgi and the nucleotide-sugar biosynthetic pathways can be directly accessed at http://cricket.ornl.gov:1555/PTR/new-image?object=SUGAR-NUCLEOTIDES.
Introduction
In recent years, the pursuit of cost-effective and sustainable methods for enzymatic degradation of plant cell wall polysaccharides to constituent simple sugars has become a major research effort (1) since such sugars can be subsequently fermented to alternative fuels. Therefore, it is important to understand how the cell walls are assembled from their basic building blocks (2), and where inside plant cells this process takes place. Secondary plant cell walls are primarily composed of a covalently cross-linked matrix of polysaccharides and lignin (2,3). Enzymatic saccharification of this matrix is impeded by a number of factors collectively contributing to the phenomenon of biomass recalcitrance (4), the resistance of plant biomass to chemical and biological catalysis of decomposition. A deep understanding of the metabolic pathways involved in cell wall assembly would be the key in identifying the factors causing biomass recalcitrance and in designing and developing rational bioengineering approaches to decrease recalcitrance of cell walls to enzymatic saccharification.
The secondary plant cell wall is a composite of polysaccharides, lignin, and proteins that is referred to collectively as lignocellulose. The wall polysaccharide fraction comprises various glycan structures, including xyloglucans, glucomannans, pectic polymers (homogalacturonan, rhamnogalacturonans RG-I, RG-II and xylogalacturononan), xylan, cellulose and callose (5). The structure and sugar composition of plant walls vary between tissues, plant developmental stage and species of plant. Across known cases the plant polysaccharides are made from nucleotide sugar precursors (2). Nucleotide sugars [nucleoside diphosphate sugars(NDP-sugars)] are composed of a nucleoside diphosphate (NDP) linked to a sugar moiety through a chemically activated phosphoester bond (6). A class of enzymes called glycosyltransferases catalyzes specific sugar incorporation from an NDP-sugar into a growing polysaccharide chain. The composition and recalcitrance of cell walls is determined in part by the availability of NDP-sugars to form different types of wall polymers. Plants produce at least 30 different NDP-sugars (7), the syntheses of which directly involve over 50 enzymes (2).
Biosynthesis of some NDP-sugars can occur across multiple cellular compartments. For example, UDP-d-xylose is synthesized from UDP-D-glucuronate both in the cytosol and in the Golgi apparatus (8), and UDP-d-glucose is synthesized from d-glucose-1-phosphate and also from sucrose in the cytosol and from d-glucose-1-phosphate in the chloroplast (9). Some NDP-sugars that are made in the cytosol, e.g. UDP-d-xylose, can be transported to the endoplasmic reticulum (ER) or the Golgi apparatus via membrane protein transporters (10,11). Synthesis of certain wall polysaccharides, e.g. xylan, xyloglucan, pectin and mannan, is believed to initiate in the Golgi, whereas cellulose synthesis occurs at the plasma membrane. Synthesis of wall glycoproteins, on the other hand, is initiated at the ER and further modified in the Golgi apparatus. NDP-sugars required for the biosynthesis of glycolipids and glycosides of secondary metabolites occur not only inside organelles but also on the surface of organelles facing the cytosol. Coordinating this extensive biochemical knowledge into a holistic understanding of the NDP-sugar biosynthetic pathways is needed in order to engineer plants with tailored glycan polymers amenable to biotechnological conversion to sugars and downstream chemical products, including biofuels. This coordination of reductionist knowledge is the essential goal of systems biology, and a key prerequisite to achieving this goal is the expression of knowledge in a form amenable to conceptual and operational modeling, and to simulation.
Computable representations are important both to the experimentalist exploring how each metabolic pathway is structured, and to the computational scientist building mathematical models to simulate the time evolution of metabolite concentrations and fluxes. A plant cell is composed of many organelles, including the nucleus, cytosol, ER, Golgi apparatus, vacuoles, lysosomes, mitochondria, chloroplasts and peroxisomes, each potentially composed of multiple distinct sub-domains and surrounded by one or more membranes. Expressing a metabolite’s production location, its concentration and its associated synthetic, transport and transforming proteins is critical to establish accurately a cellular flux network. Thus, the determination of sub-cellular localization of metabolites and enzymes participating in the biosynthetic pathways for NDP-sugars is an essential pre-requisite for tracking the metabolic fluxes to and from nucleotide sugars.
Perhaps the most mature computational system for biological representation is Pathway Tools (12,13), a software framework in which genome-scale knowledge of metabolic pathways is represented as a semantically related collection of knowledge frames (14). The central object for organism-level representation is a pathway-genome database (PGDB), which through automated methods can be generated from annotated genomes. However, incomplete genome annotation or misannotation can result in missing genes and enzymes in pathways—when many of these are missing, entire pathways can be excluded from the PGDB. Another technical issue is that the Pathway Tools framework exhibits sub-cellular compartmentalization for only transport reactions in the graphical representation of any complete pathway. Thus, a reaction in the solution phase catalyzed by a membrane-bound enzyme, for example, is not formally represented, in a pathway diagram since there is an implicit assumption that the reacting compounds and enzyme are colocated in a single compartment, the cellular interior, bounded by the cell wall. For example, the UDP-d-xylose biosynthesis in the Golgi lumen catalyzed by the Golgi membrane-bound UXS enzymes shown schematically in Figure 1 cannot be explicitly represented using the Pathway Tools framework. Unfortunately, sub-cellular compartmentalization information is generally missing from genome annotation data. Using the features of the Pathway Tools framework, enzymes and metabolites associated with any reaction could be manually assigned sub-cellular localization, in case such localization information was available. Thus, even if compartmentalization knowledge could be mapped to a representation system, this knowledge would necessarily be missing without manual curation. In order to capture localization knowledge in a PGDB constructed in an automated fashion from genome annotations, a significant degree of manual curation is therefore necessary.
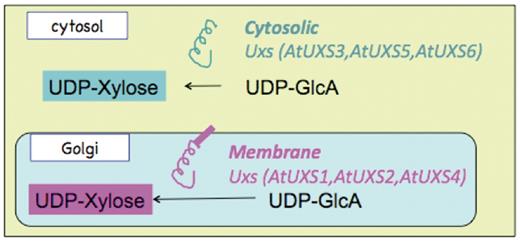
Schematic representation of the sub-cellular localization and catalytic domain orientation of cytosolic and Golgi Arabidopsis UDP-xylose synthase enzyme isoforms.
To achieve a computable representation of a subset of cell wall biosynthetic pathways with physical localization knowledge included, we first consolidated the existing knowledge on genes and enzymes involved in the NDP-sugar biosynthetic pathways in a bioenergy-relevant crop, Populus trichocarpa. We then mined from published literature and public domain databases the structure and sub-cellular localization information for these pathways and incorporated this curated information into an existing P. trichocarpa PGDB (15). In this article, we describe the curation process and adaptations needed for a multi-compartmental organism that should be easily transferable to more formal schemata, and to PGDBs of other organisms as new experimental data on those organisms become available.
Methods
Assembly and annotation of Populus NDP-sugar biosynthetic genes missing from PGDB
To distinguish between genes that are actually unknown and genes that are known but missing from the database due to incomplete/erroneous genome annotation, we first mined literature on nucleotide sugar biosynthetic pathways to identify known and unknown genes in the P. trichocarpa genome. In case of enzymes missing in Populus from a known NDP-sugar synthesis pathway, Populus open readings frames (ORFs) orthologous to genes encoding the enzymes in other organisms were identified by orthology using the INPARANOID Eukaryotic Ortholog Group Database version 7.0 (http://inparanoid.sbc.su.se/cgi-bin/index.cgi)(16) and KEGG (17–19) ortholog databases, and were suggested as the most probable candidates for the missing enzymes. Where Populus orthologs were not identified with confidence, BLAST analysis of the corresponding Arabidopsis thaliana enzyme gene, if known to exist, against the Populus genome was performed and the top among the BLAST hits with a cutoff E-value of 2 × 10−132 was considered to be the corresponding Populus enzyme.
Altering an existing PGDB to incorporate new genes and pathways
The desktop mode of Pathway Tools version 15.4 was used to modify and curate PoplarCyc 1.0 to create the prototype Populus PGDB with localization information described below. Known and inferred Populus NDP-sugar synthesis genes missing from the annotation and hence from existing pathways in the PGDB were manually incorporated using Pathway Tools editors. Pathways missing from the existing Populus PGDB (PoplarCyc 1.0) but validated experimentally were added to the prototype PGDB. New reaction frames were either created using the Pathway Tools Reaction Editor, or imported from another PGDB for each of the reactions comprising the additional pathway. These reactions were used to populate a newly created pathway frame using the Pathway Editor, and the pathway annotated using the Pathway Information Editor. An example for an added pathway was the UDP-d-galacturonate biosynthesis pathway starting from d-galacturonate, which was missing from the existing Populus PGDB. Populus enzymes in this pathway were assigned by running protein BLAST (BLASTp) on experimentally determined protein or peptide sequences involved in this pathway against the Populus proteome.
Incorporation of sub-cellular localization information on NDP-sugar biosynthesis pathways into the PGDB
The sub-cellular localization information of NDP-sugar biosynthetic pathways was either mined from literature or predicted from the amino acid sequences of the enzymes in the pathways using the publicly available software WOLF PSORT (20), SubLoc (21), PredoTar (22), MultiLoc2 (23), PredictNLS (https://rostlab.org/owiki/index.php/PredictNLS), MITOPRED (24) and CELLO (25,26), which have been summarized in Table 1. As is evident from Table 1, the MITOPRED and PredictNLS software respectively predict only nuclear and mitochondrial localization. The other sub-cellular localization programs have differential accuracy for different sub-cellular localizations. In the absence of sound experimental information, we employed all of the above programs and treated two or more consistent localization results as a consensus prediction. In the curated UDP-d-xylose biosynthesis pathway, the Populus enzymes were assumed to have the same sub-cellular localization as the lowest E-scored proteins identified in a BLASTp analysis versus Arabidopsis. The sub-cellular localization information of the Arabidopsis proteins was taken from the NCBI gene database (http://www.ncbi.nlm.nih.gov/gene/).
Comparison of sub-cellular localization prediction software used to infer sub-cellular localization of Populus enzymes involved in nucleotide-sugar biosynthesis
| Program | Prediction Method | Prediction Scope/Accuracy |
|---|---|---|
| WoLF PSORT (20) | Weighted k-nearest neighbor classifier | Predicts localization to 10 sub-cellular sites, including dual localization such as proteins which shuttle between the cytosol and nucleus; 70% accuracy for nucleus, mitochondria, cytosol, plasma membrane, extracellular and chloroplast; less accurate for peroxisome, Golgi |
| SubLoc (21) | Support Vector Machine (SVM) | 91.4% accuracy for three sub-cellular locations (cytoplasmic, periplasmic, extracellular) in prokaryotic organisms and 79.4% accuracy for four sub-cellular locations (cytoplasmic, extracellular, mitochondrial, nuclear) in eukaryotic organisms |
| PredoTar (22) | Neural Networks | Predicts sub-cellular localization of proteins to ER, mitochondria and plastids from their characteristic N-terminal targeting sequences |
| MultiLoc2 (23) | Support Vector Machine + Phylogenetic Profiles + Gene Ontology terms | High-resolution version of MultiLoc2 can predict localization to 11 eukaryotic sub-cellular locations—nucleus, cytoplasm, mitochondria, chloroplast, extracellular, plasma membrane, peroxisome, ER, Golgi apparatus, lysosome and vacuole; Accuracy—89.2% for animal proteins, 89.2% for fungal proteins and 89.4% for plant proteins |
| PredictNLS (https://rostlab.org/owiki/ index.php/PredictNLS) | Identification of sequence from protein in a carefully curated NLS (nucleotide localization signal) database | Predicts nuclear localization with close to 100% accuracy but low coverage (43%) |
| MITOPRED (24) | Identification of Pfam domain occurrence patterns and the amino acid compositional differences between mitochondrial and non-mitochondrial proteins | Predicts mitochondrial versus non-mitochondrial localization of proteins. Depending on the allowed proportions of true positives and true negatives to total positives and total negatives respectively, accuracy can vary from 71% to 92% |
| CELLO (25,26) | Two-level SVM + homology search | Predicts localization to 12 eukaryotic sub-cellular locations—nucleus, cytoplasm, cytoskeleton, mitochondria, chloroplast, extracellular, plasma membrane, peroxisome, ER, Golgi apparatus, lysosome and vacuole |
A technical issue with the Pathway Tools framework is that although compartmentalization information can be stored for metabolites participating in a reaction, the sub-cellular localization for metabolites is displayed in pathway diagrams for only transport reactions using the Cell Component Ontology (http://bioinformatics.ai.sri.com/CCO/). The latest version (15.4) of the Pathway Tools software allows the storage of compartmentalization information for any metabolite participating in a reaction via a recently added reaction frame slot called RXN-LOCATIONS. However, while the corresponding reaction page graphically displays the sub-cellular localization of the participating metabolites, such localization information is not included in the display of any pathway that includes the reaction unless it involves transport between two sub-cellular compartments. Thus, reaction of soluble metabolites in a membrane-bound organelle cannot be shown at the pathway level, and the location of soluble metabolites must be inferred by the user from the last connected transport reaction. Protein frames for enzymes and transporters, but not the compound frames for metabolites, can be manually associated with the Cellular Component Gene Ontology terms (http://www.geneontology.org) that are related to the SRI Cell Component Ontology. The graphical display page in the PGDB for any transport protein unambiguously shows the sub-cellular localization of the transporter protein. The display page for any enzyme exhibits the localization for the reaction(s) it catalyzes if such localization information is available and stored in the RXN-LOCATIONS slot of the corresponding reaction frame(s). Moreover, associated Gene Ontology terms including the Cellular Component Gene Ontology, are displayed on the PGDB pages for both enzymes and transporter proteins. In spite of all these features of the Pathway Tools framework, the graphical display of any pathway will not directly reveal the sub-cellular localization of the enzymes catalyzing the reactions constituting the pathway. To bypass such limitations in graphically displaying individual pathways, we have sought a simple mechanism for visualizing cellular compartmentalization at the pathway level.
Because explicit localization information is needed for compounds (metabolites) themselves to represent accurately a general biochemical situation, compound frames were duplicated as necessary, and renamed to include a short prefix signifying the sub-cellular localization. Using these renamed compound frames, new reaction and pathway frames were created that enabled the display of sub-cellular localization of the pathway metabolites, independently of the enzyme localization, in the graphical representation of the relevant pathways. Our current approach thus allows the pathway-level representation of metabolite compartmentalization.
Results
We have extensively curated the nucleotide sugar biosynthesis pathways in our prototype PGDB for P. trichocarpa, with special emphasis on representing the localization of enzymes in NDP-sugar synthetic pathways, and where each compound is metabolized (e.g. cytosol, Golgi). The curated nucleotide-sugar biosynthetic pathways, listed in Table 2, include most of the nucleotide-sugar biosynthetic routes from Figure 5.4 of Ref. (2).
List of curated nucleotide sugar biosynthesis pathways in the prototype Populus PGDB and the primary metabolite(s) from which these pathways are initiated
| Primary source metabolite(s) | Pathway name |
|---|---|
| d-glucose-6-phosphate | GDP-d-mannose biosynthesis |
| GDP-d-mannose | GDP-l-galactose biosynthesis |
| GDP-d-mannose | GDP-l-fucose biosynthesis I |
| l-fucose | GDP-l-fucose biosynthesis II |
| d-glucose | GDP-d-glucose biosynthesis |
| UDP-d-glucose | UDP-d-galactose biosynthesis I |
| d-galactose | UDP-d-galactose biosynthesis II |
| UDP-d-glucuronate | UDP-d-galacturonate biosynthesis I |
| d-galacturonate | UDP-d-galacturonate biosynthesis II |
| Sucrose, d-glucose-1- phosphate | UDP-d-glucose biosynthesis |
| Myo-inositol | UDP-d-glucuronate biosynthesis |
| UDP-d-glucose | UDP-d-xylose biosynthesis |
| UDP-d-xylose | UDP-l-arabinose biosynthesis I |
| l-arabinose | UDP-l-arabinose biosynthesis II |
| d-arabinose-5-phosphate | CMP-KDO biosynthesis |
| GDP-d-mannose | Ascorbate biosynthesis (l-galactose pathway) |
| d-fructose-6-phosphate | UDP-N-acetyl-d-glucosamine biosynthesis |
| UDP-d-glucose | Sucrose biosynthesis |
| UDP-d-glucuronate | UDP-d-apiose biosynthesis |
| UDP-d-glucose | UDP-sulfoquinovose biosynthesis |
| UDP-d-glucose | UDP-l-rhamnose biosynthesis |
| Sucrose, d-glucose-1- phosphate | ADP-d-glucose biosynthesis |
The detailed curation process is described for three representative pathways from the above list, in order to illustrate the process of adding new pathways and enzymes to the PGDB, and the determination, prediction and representation of sub-cellular localization of the metabolites and enzymes participating in the reactions that constitute the pathways. The three pathways considered are UDP-d-xylose biosynthesis (starting from UDP-d-glucose), UDP-d-galacturonate biosynthesis I (starting from UDP-d-glucuronate) and UDP-d-galacturonate biosynthesis II (starting from d-galacturonate).
UDP-d-xylose biosynthesis
Current experimental knowledge (6,8,27) indicates that UDP-d-xylose is synthesized from UDP-d-glucuronate in at least two separate compartments in A. thaliana, the cytosol and the Golgi lumen, as represented schematically in Figure 1. UDP-d-glucuronate is synthesized in the cytosol from UDP-d-glucose and NADH by the catalytic action of UDP-glucose 6-dehydrogenase. Any of the three cytosolic UDP-xylose synthase isozymes, AtUXS3, AtUXS5, AtUXS6, can catalyze the conversion of UDP-d-glucuronate to UDP-d-xylose in the cytosol (8). The UDP-d-glucuronate can also be transported to Golgi by a transporter (28) that is yet to be identified. In the Golgi apparatus, type II membrane-bound UDP-d-xylose synthase enzymes with catalytic portions facing the Golgi lumen (27) can convert UDP-d-glucuronate to UDP-d-xylose as represented schematically in Figure 1. However, as shown in Figure 2, the pathway representation of the same UDP-d-xylose biosynthesis pathway in the existing PoplarCyc 1.0 PGDB does not provide any information on (i) the sub-cellular localization of the metabolites that participate in the reactions constituting the pathway, (ii) the sub-cellular localization of the enzymes in the pathway and (iii) the orientation of the catalytic domains of the enzymes. For example, an enzyme bound to the Golgi membrane can have its catalytic domain in the cytosol or in the Golgi lumen. The graphical pathway representation in Figure 2 is incapable of showing the difference between these two orientations. The inability to display sub-cellular localization of metabolites in pathway diagrams by the existing Pathway Tools framework stems from the fact that in the current framework, sub-cellular localization information is displayed in the graphical representation of a complete pathway for only transport reactions in the pathway, even though such localization information can be associated with metabolites participating in any reaction. Also, the Pathway Tools framework can incorporate sub-cellular localization information about any enzyme or transporter by allowing the association of one or multiple Cellular Component Gene Ontology terms with the corresponding protein frame. However, this information is not directly evident in the representation of any pathway involving the protein as an enzyme or transporter.
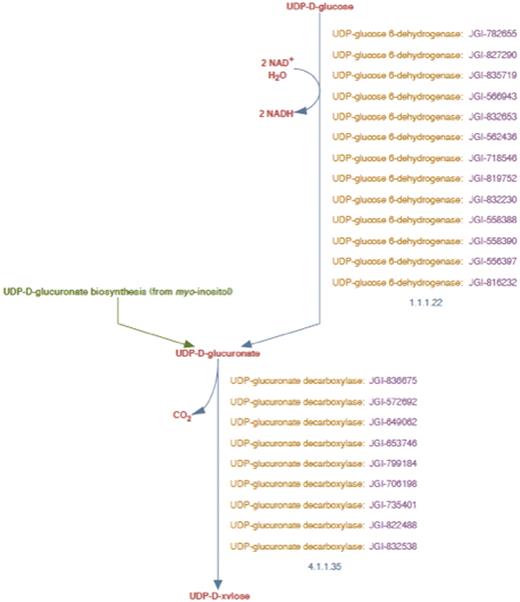
UDP-d-xylose biosynthesis pathway representation in PoplarCyc 1.0. Note that the pathway does not distinguish cytosolic reaction with EC # 4.1.1.35 from the corresponding reaction catalyzed by membrane-bound and Golgi-localized enzymes.
To capture the metabolite distributions in different locations inside the cell, the metabolite names were prepended with two-letter prefixes representing the cellular compartment containing the metabolite. Existing compound frames were duplicated and assigned names containing two-letter prefixes, which have been enumerated in Table 3. For example, new compound frames with names CY_UDP-D-xylose and GL_UDP-D-xylose were created to represent UDP-d-xylose present in the cytosol and in the Golgi lumen, respectively. Reaction frames were then constructed using these duplicated and renamed compound frames. Naturally, more detailed encoding of the sub-cellular localization in the compound frame names could be achieved simply through expansion of the lexicon in Table 3—our chosen degree of granularity simply reflected the limited extent of our present knowledge.
Two letter prefixes that identify the sub-cellular localization of metabolites
| Prefix | Cellular Compartment |
|---|---|
| CY | Cytosol |
| CS | Chloroplast stroma |
| GL | Golgi lumen |
| ER | ER |
| VC | Vacuole |
| NC | Nucleus |
| MT | Mitochondrion |
This lexicon of prefixes can be easily extended to include other cellular compartments. For example, we would use the three-letter prefix ERL to pinpoint localization to the ER lumen.
Enumeration and localization of UDP-xylose synthase isoforms
The implementation in the prototype Populus PGDB of a UDP-D-xylose biosynthesis pathway representation, that included the sub-cellular localization of the relevant enzymes, involved two main steps. First, we needed to consolidate which Populus proteins acted as enzymes for the two reactions in this pathway. In this report, we refer to different proteins in the same organism having the same enzymatic activity as isoforms. Second, we had to establish the cellular compartment to which these isoforms were localized. Finally, the isoforms were associated with reaction frames constructed from the renamed compound frames.
Reactions in the UDP-d-xylose biosynthetic pathway
The enhanced representation of the curated UDP-d-xylose biosynthesis pathway in the prototype Populus PGDB is shown in Figure 3. This pathway comprises of two reactions—UDP-d-glucuronate synthesis from UDP-d-glucose, and the synthesis of UDP-d-xylose from UDP-d-glucuronate, which are catalyzed by UDP-d-glucose 6-dehydrogenase and UDP-d-xylose synthase enzymes, respectively.
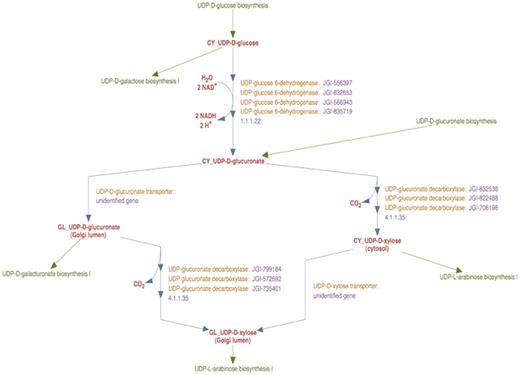
The enhanced representation of UDP-d-xylose biosynthesis pathway in prototype P. trichocarpa PGDB. In this representation the sub-cellular localization of each metabolite in the pathway is specified by a two-letter prefix (Table 3). Note that Figure 2 shows 12 genes annotated as UDP-glucose dehydrogenases and 9 genes annotated as UDP-glucuronate decarboxylase. Manual curation yields only four UDP-glucose dehydrogenase enzymes and six UDP-glucuronate decarboxylase enzymes in Populus. Also note that genes encoding Golgi-localized membrane-bound UDP-glucuronic acid decarboxylase (UXS) can now be distinguished from the same enzyme activity residing in the cytosol.
Formation of UDP-d-glucuronate from UDP-d-glucose
In the course of curating this pathway, we first considered the reaction involving the formation of UDP-d-glucuronate from UDP-d-glucose. UDP-d-glucose 6-dehydrogenase activity in plants has been reported to occur only in the cytosol (29). Moreover, the analysis of all the UDP-d-glucose 6-dehydrogenase sequences from Populus using sub-cellular localization prediction software indicated the cytosol to be the most likely target compartment. We therefore inferred that the catalytic domains of all identified Populus UDP-d-glucuronate 6-dehydrogenase enzymes are in the cytosol. A new reaction frame was created using the compound frames CY_UDP-D-glucose and CY_UDP-D-glucuronate and all the Populus UDP-d-glucose 6-dehydrogenase enzyme isoforms were associated with this reaction frame as shown in Figure 3.
Formation of UDP-d-xylose from UDP-d-glucuronate
Next we curated the representation of the synthesis of UDP-d-xylose from UDP-d-glucuronate. In plant cells, UDP-d-glucuronate can be transported from the cytosol to the Golgi lumen, as a result of which the formation of UDP-d-xylose can occur in both compartments. To describe the transport, a reaction frame was created using the compound frames CY_UDP-D-glucuronate and GL_UDP-D-glucuronate. Two separate reaction frames were created for the synthesis of UDP-d-xylose from UDP-d-glucuronate, one for the cytosolic reaction and the other for the Golgi reaction. The cytosolic reaction frame was created using the compound frames CY_UDP-D-glucuronate and CY_UDP-D-xylose and the compound frames GL_UDP-D-glucuronate and GL_UDP-D-xylose were used to populate the Golgi reaction frame.
Association of enzyme isoforms with different cellular compartments
In order to associate Populus enzyme isoforms with the cytosolic and Golgi reaction frames we had to determine which Populus isoforms were the most likely candidates to exhibit UDP-d-xylose synthase activity in the cytosol and in the Golgi lumen. As shown by Figure 2, PoplarCyc 1.0 has nine Populus UDP-xylose synthase (UXS, also known as UDP-d-glucuronate decarboxylase) enzymes that catalyze the reaction with EC # 4.1.1.35 in the UDP-d-xylose pathway. Several isoforms (AtUXS1, AtUXS2, AtUXS3, AtUXS4, AtUXS5, AtUXS6,) of the UDP-xylose synthase enzyme in A. thaliana and their sub-cellular localizations are available (8)(http://www.ncbi.nlm.nih.gov/gene/). NCBI BLASTp analysis of these Arabidopsis isoforms against all P. trichocarpa proteins in the NCBI protein database provided six significant hits with low E-values (≤2 × 10−132), all of which are assigned as Populus UXS isoforms in PoplarCyc 1.0. Each of these six proteins was found to be orthologous to one or more isoforms of the Arabidopsis UDP-d-glucuronate decarboxylase enzyme as evident from the INPARANOID database. In the absence of contradictory evidence, we considered it reasonable to assign UDP-xylose synthase activity to these six Populus proteins in the prototype Populus PGDB.
Three of these six Populus proteins with identifiers JGI-832538, JGI-822488 and JGI-706198 showed better local alignment with the shorter (∼340 aa) cytosolic isoforms of Arabidopsis UDP-glucuronate decarboxylase (AtUXS3, AtUXS5, AtUXS6), with E-values ≤ 5 × 10−180 whereas the remaining three proteins with identifiers JGI-799184, JGI-572692 and JGI-735401 aligned best (E-values ≤ 2 × 10−176) with the longer (∼430 aa) Arabidopsis type-II membrane bound UDP-d-xylose synthase isoforms that are known (27) to be localized to the Golgi lumen (AtUXS1, AtUXS2, AtUXS4). On the basis of this analysis, we inferred that Populus proteins JGI-799184, JGI-572692 and JGI-735401 were the UDP-d-xylose synthase enzyme isoforms in the Golgi lumen, whereas the Populus proteins JGI-832538, JGI-822488 and JGI-706198, were the cytosolic isoforms of UDP-d-xylose synthase. Therefore, these two sets of enzymes were associated with the Golgi lumen and cytosolic reaction frames as shown in Figure 3. In the absence of experimental knowledge about the function and sub-cellular localization of Populus UDP-d-xylose synthase proteins, our sequence homology based hypotheses would provide a starting point to curate this pathway in Poplar.
Association of ‘unidentified gene’ with observed enzymatic activity without known genetic functional annotation
A pervasive problem in genomic database curation is the existence of knowledge about enzyme activity without knowing the corresponding protein sequence or its encoding gene. For example, experimental evidence has been found that for an active transport of UDP-d-glucuronate from the cytosol into the Golgi lumen (2,28,29) but the corresponding gene has not been identified. To address this, the transport reaction frame was associated with a newly created protein frame named UDP-d-glucuronate transporter that in turn was linked to a newly created gene frame named unidentified gene. This frame, unlike other gene frames, does not represent a single gene but a class of unidentified genes. We associated this gene frame with all protein frames whose encoding genes were not identified. In the event any existing functionally unidentified ORF in the PGDB becomes identified as the UDP-d-glucuronate transporter gene, the corresponding gene frame can easily be re-associated with the UDP-d-glucuronate transporter protein frame in lieu of the unidentified gene frame, without disrupting any other relationships.
UDP-d-galacturonate biosynthesis
The second set of pathways for which we describe the enhanced representation comprises the reactions leading to UDP-d-galacturonate biosynthesis. Two separate pathways exist for the biosynthesis of UDP-GalA. Figure 4 (a) represents the UDP-d-galacturonate biosynthesis I (from UDP-d-glucuronate) pathway from PoplarCyc 1.0. This representation is lacking sub-cellular localization information about the metabolites, enzymes and enzyme catalytic domains in the pathway. Figure 4 (b) represents the second pathway, UDP-d-galacturonate biosynthesis II (from D-galacturonate) pathway, that is absent from PoplarCyc 1.0. The latter pathway (30) was only recently added to the A. thaliana PGDB AraCyc (31) version 6.0 (http://www.plantcyc.org/release_notes/release_notes_archives/aracyc/6/aracyc_release_notes.faces). Neither of these pathway representations provide any sub-cellular localization information about the metabolites involved and the enzymes that catalyze the biosynthesis of UDP-d-galacturonate, which necessitates the manual curation of both the UDP-d-galacturonate biosynthetic pathways.

(a) UDP-d-galacturonate biosynthesis I (from UDP-d-glucuronate) pathway representation in PoplarCyc 1.0. Note that no information is available to evaluate the source of UDP-glucuronate and how it becomes available to the UDP-glucuronate 4-epimerase (b) UDP-d-galacturonate biosynthesis II (from d-galacturonate) pathway representation in AraCyc 6.0.1 Note a pathway with a missing EC number for the conversion of galacturonate-1-P to UDP-galacturonate. For both parts (a) and (b), no information is available where these cellular processes occur in the cell.
UDP-d-galacturonate biosynthesis from UDP-d-glucuronate
Experimental observations indicate that UDP-d-glucuronate 4-epimerase is a type–II membrane protein with the catalytic domain facing the Golgi lumen (6). Hence, we considered it reasonable to infer that UDP-d-galacturonate biosynthesis from UDP-d-glucuronate occurred solely in the Golgi lumen. Therefore, the UDP-d-galacturonate biosynthesis pathway I was populated with a newly created reaction frame constructed using compound frames for Golgi-localized metabolites, GL_UDP-D-glucuronate and GL_UDP-D-galacturonate and all the Populus UDP-d-glucuronate 4-epimerase isoforms were associated with this particular reaction frame as shown in Figure 5.
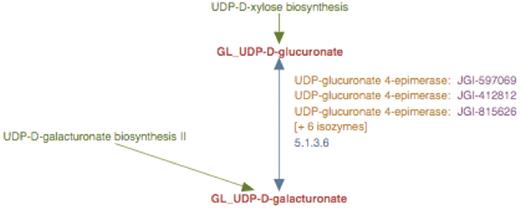
The enhanced representation of UDP-d-galacturonate biosynthesis I pathway starting from UDP-d-glucuronate in prototype Populus PGDB. The two metabolites in this pathway are linked by green arrows to two other pathways, UDP-d-xylose biosynthesis and UDP-d-galacturonate biosynthesis II.
UDP-d-galacturonate biosynthesis from d-galacturonate
Based on results from experimental studies in Arabidopsis (30) and Populus (Yang, T and Bar-Peled, M, unpublished data), we could also infer that biosynthesis of UDP-d-galacturonate from d-galacturonate occurs solely in the cytosol in two steps. First, α-d-galacturonate is phosphorylated by d-galacturonate kinase (GalAK) to yield α-d-galacturonate 1-phosphate and subsequently, α-d-galacturonate 1-phosphate undergoes uridylylation, catalyzed by SLOPPY, a non-specific UDP-sugar pyrophosphorylase, to form UDP-d-galacturonate. To represent this new pathway in the prototype Populus PGDB, we created two reaction frames, one for each of the two reactions in the cytsol and populated the first reaction frame with the compound frames, CY_D-galacturonate and CY_α-D-galacturonate 1-phosphate, and the second with the compound frames CY_α-D-galacturonate 1-phosphate and CY_UDP-D-galacturonate. A transport reaction frame to describe the transport of UDP-d-galacturonate from the cytosol to the Golgi lumen was also created using the CY_UDP-D-galacturonate and GL_UDP-D-galacturonate compound frames. These three reaction frames were used in turn to populate a newly created pathway frame for the UDP-d-galacturonate biosynthesis II pathway as shown in Figure 6.
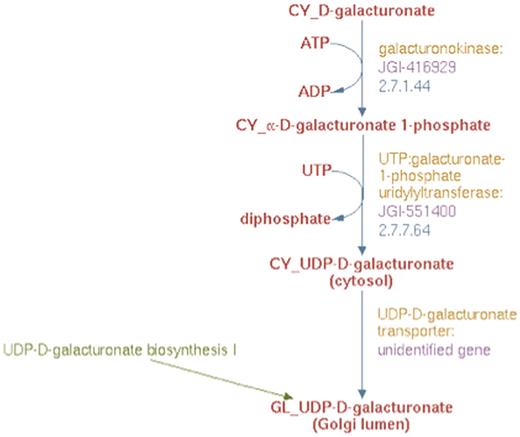
The enhanced representation of UDP-d-galacturonate biosynthesis II starting from d-galacturonate in prototype Populus PGDB.
To identify the proteins involved in this new pathway, BLAST analysis was carried out using the experimentally inferred Populus GalAK and SLOPPY protein sequences. This analysis identified the genes annotated as JGI-416929 and JGI-551400 to be encoding the GalAK and SLOPPY enzymes, respectively. These proteins and their corresponding genes were used to populate the pathway for UDP-d-galacturonate biosynthesis II as shown in Figure 6. The product of this new pathway, UDP-d-galacturonate, can be transported from the cytosol to the Golgi lumen as well (28). Since the gene encoding this transporter protein has not been identified, a newly created UDP-d-galacturonate transporter protein frame linked to the previously described hypothetical unidentified gene frame was associated with the transport reaction in the UDP-d-galacturonate biosynthesis II pathway as evident from Figure 6.
Representation of intracellular orientation of membrane proteins with multiple domains
Figure 7 represents two simple examples of membrane protein topologies (A and B). Protein A has a catalytic domain facing the Golgi lumen and Protein B, has the catalytic domain facing the cytosol. We did not attempt in this report to indicate explicitly the intracellular orientation of the multiple domains of enzymes in the enhanced pathway representations implemented in the prototype Populus PGDB; our assignment of sub-cellular localization of enzymes in pathway diagrams was intended to portray the site of catalytic activity.
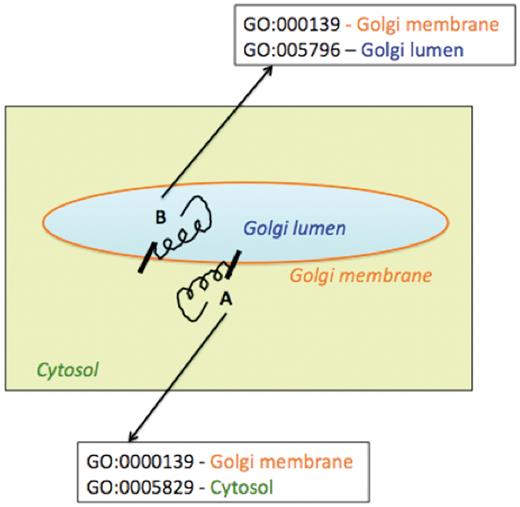
Schematic representation of association of multiple Cellular Component Gene Ontology terms with two differentially oriented Golgi membrane enzymes.
Using the existing Pathway Tools framework, we could indicate that different parts of a protein acting as an enzyme were localized to different cellular compartments by associating multiple Cellular Component Gene Ontology terms with the corresponding protein frame. To include and represent such topology information in the Pathway Tools framework, the protein frame corresponding to enzyme A would be associated with the two cellular component GO terms—GO:000139, signifying the Golgi membrane, and GO:0005829 which represents the cytosol. Similarly, the protein frame corresponding to the Golgi membrane enzyme B, with the catalytic domain facing the Golgi lumen, would be associated with the two cellular component GO terms—GO:000139 (Golgi membrane) and GO:0005796 (Golgi lumen). However such GO specifications would be evident not from the pathway representation per se, but on navigating from the pathway representation to the protein pages for the enzymes. Moreover, association of multiple cellular component GO terms would not specify which domain of the protein would be located in which cellular compartment. The explicit specification of the intracellular orientation of the catalytic or binding domains of a single polypeptide chain acting as an enzyme might be achieved by creating two separate protein frames, one for each domain, linked to the same gene frame. The two protein frames would correspond to differing sequence boundaries, different compartments and linked to a protein complex frame. Though protein complex frames are commonly used to represent complexes of multiple polypeptide chains, this approach is yet to be implemented for the different domains of a single polypeptide chain and is planned for future versions of the prototype PGDB.
Analysis of transcriptome sequencing derived single nucleotide polymorphism data using the newly curated PGDB pathways
As a case study of the utility of the newly enhanced curated nucleotide-sugar biosynthetic pathways, we analyzed transcriptome data produced in a project on improving P. trichocarpa varieties for production of biofuel (32). In the latter study twenty individual trees with different wood properties were used for xylem transcriptome analysis. Sequencing of the transcriptome revealed over 0.5 million putative single nucleotide polymorphism (SNPs) in 26,595 genes that were expressed in developing secondary xylem (wood) tissue, when compared with the reference Populus genome v2.0 from Phytozome (http://www.phytozome.net). From this genome-wide SNP data, we used the subset corresponding to the nucleotide-sugar biosynthetic genes for overlaying on the enhanced representations of the curated nucleotide-sugar biosynthetic pathways using the Pathway Tools ‘Omics Viewer’ module.
The entire transcriptome data set was retrieved from the Dryad digital repository at http://datadryad.org/handle/10255/dryad.7991 and used to calculate the total number of SNPs per gene (Supplementary Table S1). As shown in Supplementary Figure S1, the level of SNPs abundance (low, medium, high) was then estimated from the distribution of genes with different number of SNPs. We classified genes with number of SNPs from 1 to 10 as genes with low SNP abundance, from 10 to 20 as genes of medium abundance, and genes with more than 20 SNPs as genes with high SNPs abundance.
The mapping of genes with SNPs, annotated using Populus genome v2.0 to genes of the curated nucleotide-sugar biosynthetic pathways, annotated using the JGI Populus genome v1.1 is summarized in Figure 9 and Supplementary Table S3. The complete list of these genes, their SNPs abundance and the attributed pathways has been provided in Supplementary Table S4. The transcriptome data revealed that out of 172 annotated NDP-sugar biosynthetic genes (v2.0) in the curated pathways, 150 genes were transcribed in xylem tissues.

Cytosolic and Golgi isoforms of Arabidopsis and Populus UDP-xylose synthase. The cytosolic isoforms of the Arabidopsis and Populus enzymes are color coded in red and blue, respectively. The Golgi isoforms of Arabidopsis and Populus UDP-d-xylose species are color-coded in pink and orange, respectively.
Most (82%) of the transcribed 150-nt biosynthetic genes had a high or medium level of SNP abundance, i.e. more than 10 SNPs per gene, and may be under positive selection for wood properties (33–35). However, further chemical and genetic analyses are needed to firmly establish this link.
In addition, we were able to overlay SNP abundance data on individual pathway representations in the curated PGDB. Figure 9 shows the overlay of the relevant SNP data on the curated UDP-D-xylose biosynthesis pathway. All of the 10 identified genes in the UDP-D-xylose biosynthesis pathway are expressed in the xylem tissue of Populus and have high SNP abundance.
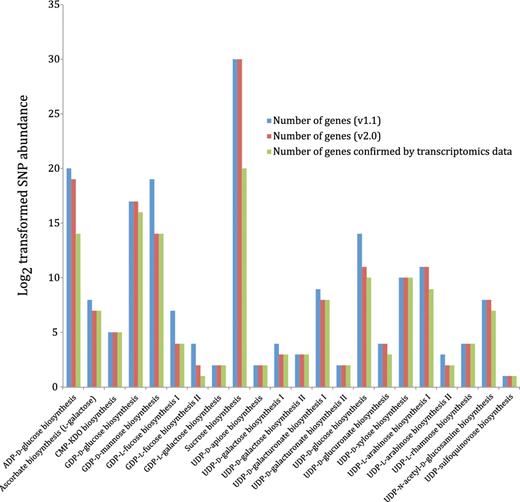
Nucleotide-sugar biosynthetic genes expressed in xylem tissue from 20 different Populus trees with different wood properties. Log2 transformed SNP abundance data for individual Populus nucleotide-sugar biosynthetic genes are overlaid with the curated nucleotide-sugar biosynthetic pathways in the prototype Populus PGDB. Genes that exhibit at least one SNP are considered expressed, whereas the status of genes lacking SNPs cannot be ascertained. UDP-L-arabinose synthesis I (from UDP-D-xylose, catalyzed by UDP-arabinose 4-epimerase) is currently represented in the PGDB as occurring in the cytosol and two different organelles—ER and Golgi, based on sequence analyses of UDP-arabinose 4-epimerase isoforms. The localization of any UDP-arabinose 4-epimerase isoform to the ER is yet to be validated by experimental data.
The overlay of SNP abundance from the Populus xylem transcriptomics dataset on the Populus cellular overview diagram with all the predicted metabolic pathways is shown in Supplementary Figure S2. It is evident from the color code on the center left of this figure that most of the carbohydrate biosynthetic reactions, that include nucleotide-sugar biosynthesis, are associated with at least one gene with high SNP abundance.
Discussion
Experimentation for decades has generated vast quantities of knowledge on nucleotide sugar biosynthesis in plants. Most of the knowledge about NDP-sugar biosynthesis in plants is embedded in natural language representations, e.g. manuscripts and technical reports and in databases such as GenBank. In both of these representations, the knowledge is presented in a form that cannot be readily incorporated into a computational model. The mapping of this knowledge to formal computable representations such as the PGDB framework is at present too complex to be fully automated. Hence, the need arises for intensive manual curation to ensure the acceptability of community knowledgebases. Although essential to retain as a thoroughly developed biological data model, knowledge representation, and accepted community standard, the existing PGDB framework is not well suited when combined with automated genome annotation pipelines to generate representations of metabolic pathways that include sub-cellular localization of the enzymes and metabolites in the pathways. This is mainly due to (i) lack of an agreed upon formal implementation for this knowledge in the Pathway Tools database schema, e.g. sub-cellular localization slots for metabolite frames, although near-term updates of the software should address this issue, (ii) unavailability of targeting information in genomic annotations and (iii) unavailability of information about protein domains (e.g. binding or catalytic) and their orientation within an organelle. The current Pathway Tools distribution allows the association of Cellular Component Gene Ontology terms with protein frames only and not with the compound frames for metabolites. Although Cellular Component GO terms can be associated with the protein frames corresponding to enzymes, such association is insufficient to pinpoint the sub-cellular localization of the different domains of the enzymes in case such domains are present in different cellular compartments.
Many proteins can have multiple sub-cellular localizations due to regulatory modifications e.g. phosphorylation and glycosylation. In the PDGDB, we include only the sub-cellular localization where a protein is functional catalytically. In other words, the specific location assigned to each enzyme in the PGDB pertains to the active form of the enzyme.
The ability to generate a pathway-level model depicting flux of precursors to the formation of a specific polysaccharide requires knowledge of compartmentalization and the component reactions of the pathway. Here, we have focused on generating a prototype PGDB for P. trichocarpa that is capable of representing sub-cellular localization knowledge about metabolic pathways by curating a well-explored subset of the metabolic pathways, the nucleotide sugar biosynthesis pathways, from an already existing P. trichocarpa PGDB. We did not aim to manually curate the entire Populus PGDB. The sole purpose of our curation effort was to outline how we can establish a prototype platform by utilizing the existing Pathway Tools framework, originally developed for prokaryotic organisms, to capture and visualize eukaryotic data on pathway topology that includes sub-cellular localization information. The sub-cellular localizations of enzymes in the nucleotide-sugar biosynthetic pathways in the current version of the PGDB are mostly predicted from sequence analysis using commonly available bioinformatics software. In case new experimental evidence on nucleotide-sugar biosynthesis becomes available that refutes the predicted results, the database will be manually curated to keep it up to date with the most recent experimental findings.
The current state of knowledge on nucleotide sugar biosynthesis in plants is largely a collage of experimentally derived information extracted from different plant species. Thus, there are several factors that hinder the consolidation of this knowledge for a particular plant species. For example, not all plants have the same nucleotide sugar biosynthetic pathways. For example, the green alga Chlamydomonas reinhardtii has a UDP-d-galactose mutase which, based on sequencing data, is absent in land plants. Also, the same reaction, e.g. UDP-d-glucuronate → UDP-d-xylose, in multiple plant species can be catalyzed by different proteins that might reside in different cellular compartments. Figure 10 shows the different isoforms of Populus and Arabidopsis UDP-d-xylose synthase. Moreover, the same metabolite, e.g. UDP-d-glucose, can be produced by different metabolic routes in different plants. Here, we have outlined possible bioinformatics workflows to consolidate pathway information in one species (P. trichocarpa) based on experimentally derived knowledge available for other plant species (A. thaliana).
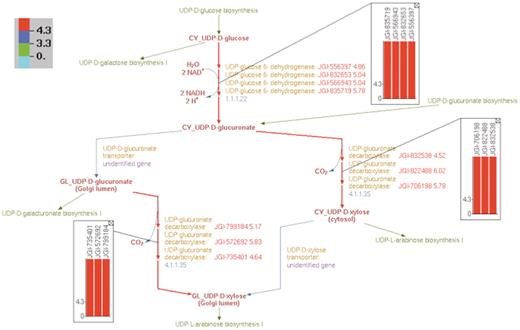
Overlay of log2-transformed SNP abundance for genes in the UDP-d-xylose biosynthesis pathway. Individual reaction representations are highlighted using the color code at the top left corner. The numbers in the color code define the ranges of the log2(SNP abundance) values of the genes in such a way that red, blue and green colors correspond to genes with high, medium and low SNP values, respectively. The reaction arrows for UDP-d-glucuronate formation, and for both the cytosolic and intra-Golgi conversions of UDP-d-glucuronate to UDP-d-xylose are highlighted in red since all the corresponding genes have high log2(SNP abundance). The color-coded log2(SNP abundance) values for the transcriptome genes encoding the enzymes catalyzing each reaction can be exhibited using pop-up bar diagrams, shown as inset figures. The absolute log2(SNP abundance) values for the genes that explicitly appear on the pathway diagram occur next to the corresponding gene locus tags and are color coded as well.
Accurate capture and representation of metabolic pathway knowledge is one of the pre-requisites for developing accurate metabolic pathway models. The current work shows that the existing Pathway Tools framework can be used to enhance significantly the capture and representation of eukaryotic metabolic pathway knowledge. New pathway knowledge acquired by additional gene calling based on published literature and recent experimental studies was thereby easily incorporated, and replication and appropriate renaming of compound frames in the Pathway Tools framework allowed the specification and representation of sub-cellular localization of pathways. In this light, it should be mentioned that the biggest challenge in the representation of the sub-cellular localization of eukaryotic metabolic pathways lies in acquiring the knowledge of sub-cellular compartmentalization of eukaryotic metabolic pathways.
The current scheme presents some challenges a formal schema that included compartmentalization information in compound frames would surmount. By replicating compound frames and creating differing but related identifiers, queries can become more complex—if all pathways containing a certain compound were desired, regular expressions might have to be used to discount the prefix, for example. Such complexity (or kludginess, depending on perspective) can be hidden from the user through dialog-based queries. Also, because different compound frames were intended to encode different chemical structures and not the same structure in different physical cellular locations, processes that depend subtly on the originally intended data model could become broken. However, the current scheme offers two advantages. First, it is lightweight in the sense of requiring neither the modification of the underlying Pathway Tools data model or schema, nor programming of the Pathway Tools software framework. Second, translation of a PGDB to a future version with an explicit localization slot in any compound frame should be fairly trivial, by collapsing the multiple prefixed identifiers for a given compound back to a single frame with a ‘compartment’ slot filled in with the appropriate term.
An elegant way of representing metabolite localization in the pathway would necessitate programming of the Pathway Tools framework so that metabolite localization information stored in the existing RXN-LOCATIONS slots of reaction frames can be transmitted to the graphical representation of related pathways. An alternate way to represent the sub-cellular localization of enzymes in individual pathways would involve adding an attribute to individual enzyme names in pathway diagrams, such that the attribute will indicate the localization of the different domains of the enzyme. However, the implementation of this more detailed way of representing enzyme localization would involve extending the Pathway Tools database schema by introducing slots or frames for individual protein domains, as well as programming of the Pathway Tools framework that will enable the transmission of the information from these new slots or frames to the visual representation of pathways. These modifications would need to be done by developers with access to source code, and who can implement such a scheme without corruption of the data model. We expect future versions of the Pathway Tools software to implement the display of subcellular localization in individual pathway in a more efficient fashion without frame duplications.
Two important factors besides the risk of corrupting the Pathway Tools data model have led us to avoid pursuing the representation of enzyme localization at the level of individual domains, which would permit a finer-grained understanding of the associated reaction. First, we believe this granularity to be too fine for pathway-level display. Although we did wish to make the display more detailed than the Pathway Tools default with no compartmentalization, we do not wish to confuse the user of the PGDB by including too much information. Domain-level compartmentalization would be an excellent addition for the datasheet about the protein itself; however, at the pathway level, we feel it sufficient to specify the compartment where the chemistry of the reaction occurs—this, together with metabolite compartmentalization information, permits a user to infer, for example, whether a transport event is needed for the reaction to occur. A second factor preventing our inclusion of domain-level compartmentalization is the general lack of knowledge on this topic. However, future extension for genome-scale databases to include domain-level enzyme compartmentalization will be valuable as such knowledge accumulates.
It should be noted that nothing in the informal workflow outlined here is specific to P. trichocarpa. Hence, this approach might be useful in deriving compartmentalization and in filling pathway holes found in other target organisms. An intriguing possibility is an automated framework that would both mine knowledge of eukaryotic metabolic pathway sub-cellular localization and construct the appropriate representations, such as was done by hand here. It would be possible in principle, although probably very challenging in practice, to generate such a framework by interfacing automated PGDB construction with software for extracting localization information already included in publicly available databases, natural language analysis of a targeted corpus of scientific literature, and sub-cellular localization prediction. Once a critical mass of more highly curated PGDBs are available, such a scheme could be augmented with analysis and transfer of appropriate pathway frames incorporating sub-cellular localization information to PGDBs of other plants during post-annotation automated generation.
In summary, we have devised a simple means of encoding sub-cellular localization into the existing Pathway Tools framework that permits pathway-level representation of metabolite and protein compartmentalization directly, and facile encoding of the present state of knowledge regarding nucleotide sugar biosynthesis pathways in plants, specifically P. trichocarpa. The best existing Poplar PGDB was curated to include knowledge of the pathway biochemistry, as well as the physical localization of the pathway reactions. The resulting database, captured in a community-standard framework, permits broad access to this information through both a Web browser interface, and through the extensively developed visualization and editing tools provided as part of the Pathway Tools platform. In particular, the prototype database was able to be deployed immediately to an interested community of biofuels researchers. Adherence to this platform’s standards also provides access to the aggregate biochemical representation experience and formal data schemata already included, as well as any new developments to be added to Pathway Tools in the future.
In addition, we have provided an example of how experimental ‘omics’ data can be overlaid on pathway representations in the prototype Populus PGDB. Overlay of transcriptome-sequencing derived SNP abundance data from a recently obtained Populus xylem transcriptomics dataset on the UDP-D-xylose biosynthesis pathway indicates that all of the genes in this pathway are confirmed by the transcriptome and have high SNP abundance.
The prototype PGDB described can serve as a reference representation for biochemical kinetic model construction, simulation of which can aid in devising cell wall bioengineering strategies. Key research areas include metabolic control of NDP-sugar biosynthesis in Populus, and potential upstream transport limitations to NDP-sugar incorporation in the cell wall. Greater appreciation of these pathways’ operation will enable the rational redesign of NDP-sugar metabolism to tailor cell wall compositions in Populus in search of an improved biofuel feedstock with less recalcitrant cell walls.
Funding
National Science Foundation (Grant IOB-0453664) (M.B.-P.); BioEnergy Science Center (Grant DE-PS02-06ER64304); Plant Microbe Interfaces Scientific Focus Area (Grant DE-AC05-00OR22725) that are supported by the Office of Biological and Environmental Research in the DOE Office of Science. Funding for open access charge: XXX.
Conflict of interest. None declared.
Acknowledgements
The authors would like to thank Dr Peter Karp (SRI) for critically reading the manuscript and Drs Brian H. Davison (ORNL), Debra Mohnen (CCRC) and Wesley B. Jones (NREL) for their efforts to initiate and organize this multi-institution collaboration. The authors also thank Dr Ting Yang (CCRC) for providing sequence data, Priya Ranjan (ORNL) for providing a table for mapping of the P. trichocarpa genome annotation from v2.0 to v1.1, and Denise Hummel and Bill Van Meter (NREL) and SRI support staff for assistance with Pathway Tools software.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}