





Abstract
The increased throughput and decreased cost of next-generation sequencing (NGS) have shifted the bottleneck genomic research from sequencing to annotation, analysis and accessibility. This is particularly challenging for research communities working on organisms that lack the basic infrastructure of a sequenced genome, or an efficient way to utilize whatever sequence data may be available. Here we present a new database, the Assembled Searchable Giant Arthropod Read Database (ASGARD). This database is a repository and search engine for transcriptomic data from arthropods that are of high interest to multiple research communities but currently lack sequenced genomes. We demonstrate the functionality and utility of ASGARD using de novo assembled transcriptomes from the milkweed bug Oncopeltus fasciatus, the cricket Gryllus bimaculatus and the amphipod crustacean Parhyale hawaiensis. We have annotated these transcriptomes to assign putative orthology, coding region determination, protein domain identification and Gene Ontology (GO) term annotation to all possible assembly products. ASGARD allows users to search all assemblies by orthology annotation, GO term annotation or Basic Local Alignment Search Tool. User-friendly features of ASGARD include search term auto-completion suggestions based on database content, the ability to download assembly product sequences in FASTA format, direct links to NCBI data for predicted orthologs and graphical representation of the location of protein domains and matches to similar sequences from the NCBI non-redundant database. ASGARD will be a useful repository for transcriptome data from future NGS studies on these and other emerging model arthropods, regardless of sequencing platform, assembly or annotation status. This database thus provides easy, one-stop access to multi-species annotated transcriptome information. We anticipate that this database will be useful for members of multiple research communities, including developmental biology, physiology, evolutionary biology, ecology, comparative genomics and phylogenomics.
Database URL:asgard.rc.fas.harvard.edu
Introduction
In the early ‘genomic era’ of the late 1990s and early 2000s, the genomes of several long-standing traditional laboratory model organisms were completely sequenced (1–5), which galvanized their respective fields by offering enormous amounts of new data for analysis. Importantly, the beneficial effects of these genome projects were maximized by the simultaneous creation of dedicated web interfaces (e.g. 6–11), or incorporation of the data into existing community databases (e.g. 12), so that users could immediately and easily access and search genome sequences. The advent of next-generation sequencing (NGS) has further advanced biological research not only in traditional model systems, but also in an increasing number of clades that previously lacked genomic data (13–22). High-throughput NGS technology now enables researchers studying non-traditional model organisms to obtain genomic or transcriptomic data relatively efficiently and at modest costs.
Transcriptome and RNA-Seq data are currently the fastest growing category of genomic data across many biological research fields (23, 24). However, unlike the pioneering genome sequence projects, these smaller ‘omics’ datasets are usually minimally annotated to meet the needs of a specific research goal, and are rarely available or searchable in assembled or annotated form. The NCBI’s Sequence Read Archive (SRA) (25) provides a means of archiving data obtained from 454 pyrosequencing, Illumina Genome Analyzer sequencing and other NGS platforms. However, it does not allow for deposition or searching of assembled transcriptomes. Basic Local Alignment Search Tool (BLAST) searches of the SRA data are possible, but only by selecting a single SRA dataset for a given organism at a time. The commonly used NCBI BLAST portal (http://blast.ncbi.nlm.nih.gov/Blast.cgi) does not include SRA data within the nucleotide collection or reference RNA sequences (refseq_rna), although it does allow SRA searches as a specialized BLAST option. The transcriptome shotgun assembly (TSA) database (http://www.ncbi.nlm.nih.gov/genbank/tsa/) allows storage of complete assemblies, but annotation of deposited assemblies is not required. As a result, the potential for leveraging the vast majority of transcriptome data generated are diminished.
One animal clade for which substantial amounts of NGS data are being generated is the Arthropoda (e.g. 21, 22, 26–34). The most speciose animal phylum, arthropods include spiders and scorpions (Chelicerata), centipedes and millipedes (Myriapoda) and insects and crustaceans (Pancrustacea). Arthropods have served as important models for studies of evolutionary biology (35–39), ecology (40–42), physiology (43, 44) and biomechanics (45, 46). As prevalent vectors of human disease and major agricultural pests, arthropods are also of significance to economic development and global health. Finally, the fruit fly Drosophila melanogaster has been a powerful model organism for the study of genetic, evolutionary, developmental and biomedical research for over a century (47, 48) and possesses the most sophisticated genetic analysis toolkit of any animal model (12, 49–51). As a result, functional genetic and genomic studies in other arthropods have flourished by taking advantage of the well characterized Drosophila genome as a point of reference (9, 11, 52–54). However, these studies exhibit a distinct phylogenetic bias: the vast majority of arthropod genomic data available have been generated for the holometabolous insects, which undergo complete metamorphosis. Because the Holometabola are derived in many respects compared with the basally branching Hemimetabola (insects that do not undergo metamorphosis) and other arthropods (55), many recent efforts have used NGS to obtain transcriptome data from other emerging model arthropods (19, 21, 22, 56, V. Zeng, B. Ewen Campben, H.W. Horch et al., submitted for publication). These projects are particularly important for new model organisms for which functional genetic techniques have been developed, as the roles of genes discovered through NGS can be functionally tested in these animals. However, even if these data are deposited in the SRA, as described above, there is typically no public access provided to search the annotated data.
To address this problem, we have created a searchable database of the annotated transcriptomes of three emerging model arthropods, which provide data for a range of phylogenetic diversity within Pancrustacea. All of these organisms have risen to prominence as emerging model organisms due to their ease of inbred laboratory cultures, year-round embryo collection and gene expression analysis via in situ hybridization and antibody staining. The milkweed bug Oncopeltus fasciatus (Figure1, left) belongs to the order Hemiptera, the sister order to all holometabolous insects including Drosophila (55). Determination of gene function is possible in O. fasciatus using maternal or embryonic RNA interference (RNAi) (57–61). The amphipod crustacean Parhyale hawaiensis (Figure 1, middle) is a member of the crustacean class Malacostraca and thus serves as a Pancrustacean outgroup to insects (62). Multiple functional genetic tools have been developed for P. hawaiensis, including gene knockdown by small interfering RNAs (siRNAs) and morpholinos (63–65), stable germ line transgenesis (66), inducible gene overexpression (67), site-directed insertions and enhancer trapping (68). The cricket Gryllus bimaculatus (Figure 1, right) branches basally to both Holometabola and Hemiptera and has multiple advanced functional genetic techniques available, including maternal, zygotic, nymphal and regenerative RNAi (69–72), stable germ line transgenesis (73) and targeted genome editing (74).
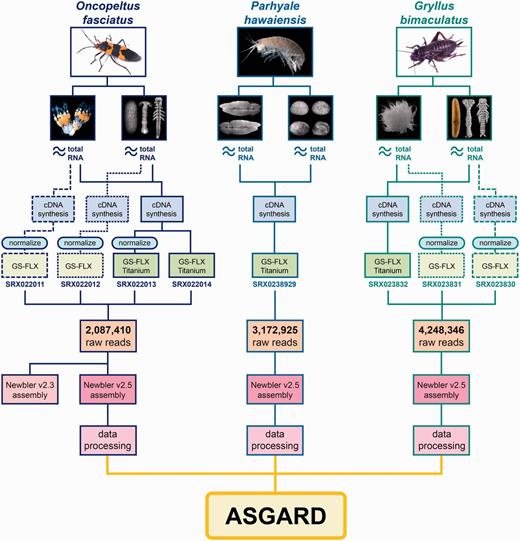
Origin and processing of data contained in ASGARD. Flowchart showing adult specimens and tissue types obtained for ASGARD v1.0 organisms O. fasciatus, P. hawaiensis and G. bimaculatus. Total RNA was prepared separately from ovaries and mixed-stage embryos and used for cDNA synthesis. For insect samples, some libraries were normalized in pilot experiments and sequenced using GS-FLX 454 pyrosequencing. The majority of reads used in the de novo assemblies were obtained using GS-FLX Titanium 454 pyrosequencing. SRA accession numbers are shown for each sequenced sample. Reads from each organism were pooled, assembled with Newbler v2.5 and annotated using the data processing pipeline described in the main text. The resulting data are searchable via the ASGARD web interface.
The database presented here provides a way for researchers in any field to easily search for genes of interest in these animals among previously described maternal and embryonic transcriptome data (21, 22, V. Zeng, B. Ewen Campben, H.W. Horch et al., submitted for publication). The database provides BLAST search capability for any or all transcriptome assemblies, something which is not possible with SRA BLAST searches as the SRA houses only unassembled, un-annotated raw reads. Moreover, all transcriptomes have been annotated for gene orthology, protein coding regions, functional protein domains and Gene Ontology (GO) terms, allowing researchers to search for genes of interest using any of these identifiers.
Database content
The ASGARD integrates annotated assembly information from the maternal and developmental transcriptomes of O. fasciatus, P. hawaiensis and G. bimaculatus. Full details of the creation, assembly and annotation of each transcriptome have been previously described (21, 22, V. Zeng, B. Ewen Campben, H.W. Horch et al., submitted for publication). Briefly, the transcriptome for each organism was created by isolating total RNA from adult ovaries and from embryos of multiple developmental stages (Figure 1, blue boxes). The complementary DNA (cDNA) libraries were sequenced using 454 GS-FLX and Titanium pyrosequencing (Figure 1, green boxes) and assembled using Newbler v2.5. In the cases of the two insects, pilot experiments using GS-FLX and/or library normalization were carried out in the course of transcriptome optimization (Figure 1, blue boxes); the data in ASGARD contains the results of all such pilot experiments incorporated into the relevant assembly (21, V. Zeng, B. Ewen Campben, H.W. Horch et al., submitted for publication). In the case of O. fasciatus, the initial assembly was performed with Newbler v2.3 (21), but prior to integration into ASGARD the raw reads were re-assembled with Newbler v2.5 to make the assembly comparable with those of P. hawaiensis (22) and G. bimaculatus (V. Zeng, B. Ewen Campben, H.W. Horch et al., submitted for publication) (Table 1).
Transcriptome assembly statistics for data contained in ASGARD V1.0
| Species | O. fasciatus | P. hawaiensis | G. bimaculatus | |
|---|---|---|---|---|
| Class, order | Insecta, Hemiptera | Malacostraca, Amphipoda | Insecta, Orthoptera | |
| No. of raw reads | 2 087 410 | 3 172 925 | 4 248 346 | |
| Mean read length | 297 | 400 | 349 | |
| No. of raw base pairs (bp) | 619 186 225 | 1 204 620 614 | 1 483 726 666 | |
| Assembler(s) used | Newbler v2.3, CAP3 | Newbler v2.5 | Newbler v2.5 | Newbler v2.5 |
| No. of reads input for assembly (percent of raw reads) | 2 041 966 (97.8) | 2 041 951 (97.8) | 3 157 373 (99.5) | 4 216 721 |
| No. of base pair input for assembly (percent of raw base pairs) | 566 097 669 (91.4) | 566 080 984 (91.4) | 1 179 544 291 (97.9) | 1 449 059 795 (97.7) |
| No. of reads used in assembly products (percent of reads input) | 1 773 450 (86.8) | 1 794 099 (87.9) | 2 625 830 (83.2) | 4 146 625 (98.3) |
| No. of base pairs assembled (percent of base pair input) | 508 738 047 (89.9) | 509 976 789 (90.1) | 1 027 860 567 (87.1) | 1 383 106 269 (95.4) |
| No. of isotigs | 21 097 | 20 985 | 35 301 | 21 512 |
| No. of isogroups | 16 617 | 16 849 | 25 735 | 16 456 |
| No. of singletons (percent assembled reads) | 178 770a (10.1) | 168 807a (9.5) | 276 564 (8.8) | 120 805 (2.9) |
| No. of CAP3 contigs | 28 143 | 29 434 | n/ab | n/a |
| Isotig N50 | 1735 | 1651 | 1510 | 2133 |
| No. of isotigs with BLAST hitsc (%) | 7219d (43.4) | 7305d (43.4e) | 10 424 (29.5%) | 11 943 (55.5) |
| No. of singletons with BLAST hits (%) | 2367f (2.8) | 2350g (2.8) | 9583 (3.5) | 10 815 (9.0) |
| No. of unique BLAST hits | 10 775 | 10 886 | 19 067 | 19 874 |
| No. of CAP3 contigs with BLAST hits (%) | 2594 (9.2) | 2642 (9.0%) | n/a | n/a |
| Mean no. of contigs per isotig | 1.9 | 1.8 | 2.1 | 1.7 |
| Mean no. of isotigs per isogroup | 1.3 | 1.2 | 1.4 | 1.2 |
| Mean coverage per base pair | 23.2 | 28.7 | 25.4 | 51.3 |
aRefers to the number of singletons produced by the Newbler v2.3 assembly. These singletons were further assembled with CAP3, resulting in CAP3 contigs and CAP3 singlets (reads still left as singletons); bCAP3 assembly of Newbler singletons was not performed for P. hawaiensis or G. bimaculatus assemblies; cBLASTx performed against the NCBI non-redundant database (nr) with E-value cutoff of e−5, unless otherwise specified; dBLASTx performed against RefSeq Protein database; ePercent isotigs with BLAST hits calculated by dividing by the number of isogroups, because in this case, only one isotig per isogroup was used for BLAST analysis; fTotal number of CAP3 singlets following CAP3 assembly of Newbler v2.3 singletons was 84 388; gTotal number of CAP3 singlets following CAP3 assembly of Newbler v2.5 singletons was 85 053.
The initial descriptions of the O. fasciatus and P. hawaiensis transcriptomes included only BLAST-based and manual gene annotation (21, 22). For all transcriptomes, ‘significant’ BLAST hits were considered as those with a top hit meeting an E-value cutoff of 1e−5 unless otherwise indicated. To improve utility of these data in preparation for ASGARD deposition, the O. fasciatus and P. hawaiensis transcriptomes were further annotated to match the annotation status of the G. bimaculatus transcriptome (V. Zeng, B. Ewen Campben, H.W. Horch et al., submitted for publication) by using: (i) an automated tool called ‘Gene Predictor’ that determines putative orthology based on the best reciprocal top BLAST hit against the D. melanogaster proteome (V. Zeng, B. Ewen Campben, H.W. Horch et al., submitted for publication); (ii) Expressed Sequence Tag (EST) Scan (75) to detect putative coding regions for all predicted transcripts; (iii) InterPro Scan (76) to detect functional protein domains for all predicted protein-coding transcripts and (iv) GO terms (77) obtained by assigning each transcript the GO term of the best reciprocal BLAST hit from the D. melanogaster proteome as in (i), or in the absence of such a hit, the GO term of the top BLAST hit from the NCBI non-redundant database (nr). In total, ASGARD contains data derived from annotating the assembly products of 9 508 681 raw 454 pyrosequenced reads (Figure 1, orange boxes) totaling over 3.25 billion base pairs (Figure 1, Table 1). The outputs of the Newbler assembly contained in ASGARD include ‘isotigs’ (continuous paths through a given set of contigs, named ‘isotigXXXXX’ where XXXXX is a five-digit unique numeric identifier) and ‘singletons’ (high quality single reads lacking significant overlap with any other read, named with a 14-character unique identifier). Newbler also predicts ‘isogroups’, which are groups of isotigs assembled from the same set of ‘contigs’ (groups of reads with significant overlapping regions). However, because of the limitations inherent in making genome structure predictions based on de novo transcriptome data alone [discussed previously (22, V. Zeng, B. Ewen Campben, H.W. Horch et al., submitted for publication)], ASGARD makes no assumptions about putative gene numbers of any component organisms and does not contain explicit annotation of isogroups. The assembly and annotation of all raw data yielded information on 77 798 putative transcripts (isotigs), 59 040 putative genes (isogroups) and 566 176 singletons (unassembled high-quality reads) that obtain 49 827 unique BLAST hits in nr (Figure 1, pink boxes).
We designed ASGARD to serve two principal purposes: (i) to provide a centralized repository for these and future assembled and annotated transcriptomes from emerging model arthropods, as distinct from the source of raw reads already available from the SRA and (ii) to allow users to search for genes of interest in any or all transcriptomes, based on sequence similarity, putative orthology or predicted functional criteria. In this way, ASGARD can help researchers from any field of biology that need sequence data from these arthropods. The following sections briefly describe the main annotation strategies used to provide the data for ASGARD, full methods of which are described elsewhere (21, 22, V. Zeng, B. Ewen Campben, H.W. Horch et al., submitted for publication).
Coding region predictions
Regardless of whether an isotig or singleton (assembly product) obtained a significant BLAST hit, the predicted coding region of each assembly product was processed by EST Scan (75). EST Scan performs coding region prediction based on a Markov model of protein coding sequences to differentiate untranslated regions (UTRs), including 3′- and 5′-UTRs, from coding regions. This probability model is also useful in detecting sequencing errors often associated with the 454 pyrosequencing platform, including the difficulty in resolving homopolymer repeats that may generate frame shifts in the translated protein (78). The transcript position of the highest scoring predicted coding region generated by EST Scan is recorded in the database, which also provides information regarding whether the assembly product likely represents the positive or the negative strand of the actual transcript. This information is visually represented with a schematic diagram in ASGARD. This analysis can thus provide users with putative coding region information for all assembly products of the transcriptomes, even if an isotig or singleton has no predicted orthology to known sequences.
Protein domain predictions
For those assembly products with detected coding regions, their predicted proteins were further annotated using InterPro Scan (76). This tool searches for motif signatures of known functional protein domains within the predicted coding regions of assembly products. To encompass the widest possible range of methods of defining protein motifs, several different protein motif databases are used for this annotation, including ProDom (79), PRINTS (80), SMART (81), TIGRFAMs (82), Pfam (83), Prosite (84), PIRSF (85), SUPERFAMILY (86), CATH (87), PANTHER (88), SignalPHMM (89) and Transmembrane (90). The location of predicted protein domain motifs within the translation is displayed schematically, enabling ASGARD users to better interpret the potential structure and functions of predicted proteins. A link to the relevant protein database website is also generated for each predicted motif, so that users may easily obtain details of specific protein domains.
Orthology (gene identity) predictions
Assembly products of transcriptomes were compared with the NCBI nr database to determine their similarity to known sequences, and the top 50 BLAST hits meeting an E-value cutoff of 1e−5 were recorded in the database. The criterion of reciprocal best BLAST hit against the D. melanogaster proteome is a commonly used method of automated annotation in projects involving insect genomes (e.g. 91, 92). We therefore additionally employed this method of putative orthology assignment as the D. melanogaster proteome is well annotated, and is the best annotated arthropod proteome derived from a complete genome sequence. To do this, we used a previously described custom script called ‘Gene Predictor’ (V. Zeng, B. Ewen Campben, H.W. Horch et al., submitted for publication). Specifically, each D. melanogaster protein was queried against each assembly product of the ASGARD BLAST databases using tBLASTn and conversely, each assembly product was queried against the D. melanogaster proteome BLAST database using BLASTx (93). Because each predicted transcript may comprise multiple assembly products and multiple predicted isoforms may exist for a transcript as discussed below (see ‘Treatment of putative paralogs, isoforms and singletons’ section), only the top 50 results of the D. melanogaster proteome against assembly product query were retained. Similarly, only the top BLAST result of each assembly product against the D. melanogaster proteome was used to infer whether a specific D. melanogaster protein was the best match for a given assembly product. To prevent a given assembly product from being annotated repeatedly as different isoforms of a single D. melanogaster gene, only the longest D. melanogaster protein isoform was considered. To prevent an assembly product from escaping annotation if its top BLAST hit was not the longest isoform of a D. melanogaster gene, all D. melanogaster protein hits were verified based on the gene rather than a particular protein product.
Treatment of putative paralogs, isoforms and singletons
During iteration through the top 50 BLAST results of the D. melanogaster proteome against the assembly products, we also assessed whether those hits that were assigned a putative orthology based on reciprocal BLAST (which we call here ‘verified’ by Gene Predictor) might be paralogs or isoforms. We used criteria for assessing putative paralogs as previously described for the initial assemblies of ASGARD transcriptomes (21, 22, V. Zeng, B. Ewen Campben, H.W. Horch et al., submitted for publication). Specifically, we asked whether the alignments of verified assembly products overlapped with any other verified assembly product that had been assigned the same orthology. If the sequences did not overlap, they were considered to be fragments of a single isoform that were not assembled together due to insufficient overlap. If the assembly product sequences overlapped, then we asked whether they had been predicted as isoforms based on the original assembly. Briefly, if two isotigs shared a contig, they were considered likely to belong to the same isogroup, suggesting that they were isoforms of the same gene. Singletons lack isogroup information and were thus considered as putative isoforms. If overlapping assembly products were not predicted to belong to the same isoform, they were considered as putative paralogs. The same method of paralog inference was used to determine whether specific D. melanogaster genes might have multiple paralogs among the assembly products.
We emphasize that although accurate prediction of isoforms, paralogs and orthologs is not possible in the absence of complete genome sequences and phylogenetic analysis, the assumptions described above will not prevent ASGARD users from obtaining meaningful biological information from the database. Our aim is to facilitate annotation of assembly products, allowing users to retrieve sequence data from these emerging model organisms based on similarity to known genes or predicted function. Even if not all paralogy or isoform assignments of assembly products are accurate, sequence similarity can still be revealed by the reciprocal BLAST searches performed by Gene Prediction.
GO annotations
The GO annotation of each assembly product was obtained using blast2go (94, 95). The top 50 BLASTx hits of each assembly product against nr were exported in M7 format (XML). These XML BLAST data were then processed using the command-line version of blast2go (b2gPipe) with database version 2.3.5.
Database implementation
Unlike a genome database, the database schema of ASGARD is designed around isotigs and singletons (assembly products) rather than genome scaffolds. Figure 2 shows a schematic representation of the database schema with relevant input data and user interface outputs. Because all singletons and isotigs obtained from assembly of raw sequence data have unique identifiers, the ASGARD database uses these identifiers (called ‘read name’ or ‘Sequence ID’) to associate each assembly product with all annotation data. Singular data (including read name, isogroup, assembly product length, nucleotide sequence, predicted coding sequence, translation and coding strand) are stored within one central table (Figure 2, ‘assembled_sequences’). Data where multiple values exist for each assembly product (including protein motifs, BLAST hits and GO annotations) are stored in separate tables (Figure 2, ‘protein_feature’, ‘blast_annotation’ and ‘go_annotation’) with multi-key indexing associated with the read name of the central table. Lastly, data produced by Gene Predictor are stored in a separate table (Figure 2, ‘gene_prediction’). Because under our annotation conditions every assembly product can only be the ortholog of a single gene, the gene prediction table is uniquely keyed to each assembly product identifier. This table is also multi-key indexed for the predicted orthologous gene, which allows rapid query of all assembly products annotated as putative orthologs of that gene.
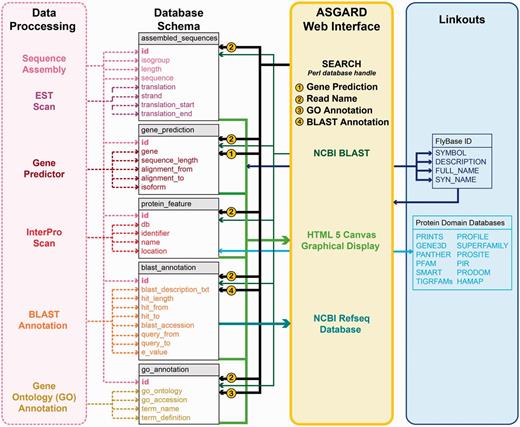
ASGARD database schema. Schematic of database implementation showing destination tables (gray/white) for each data type created by the data processing pipeline (pink), how users may access those data via the ASGARD web interface (yellow) and sources of linkout data provided by the ASGARD search results displays (blue). See main text for details.
To develop ASGARD and implement the ASGARD schema, we used MySQL, custom Perl scripts with the Apache web server hosting tool, and developed custom HTML5 and JavaScript rendering code for the visual output displays.
Site navigation and overview of search capabilities
The home page of ASGARD provides a brief description of the provenance and preparation of the transcript sequences house in the database. To the bottom left is a ‘News’ area where ASGARD development updates are posted. To the bottom right is a ‘Publications’ area that provides updated literature obtained from PubMed mentioning any of the ASGARD transcriptome organisms. A navigation menu at the top of the page allows users to: (i) access pages where they can search ASGARD annotations; (ii) use BLAST to query transcriptome sequences; (iii) obtain contact information for ASGARD developers; (iv) obtain citation information for ASGARD and its component transcriptomes, including links to the PubMed entries and PDFs for download and (v) download the assemblies of all three transcriptomes in FASTA format. The download page also provides links to previously described custom scripts used for assembly (21, 22, V. Zeng, B. Ewen Campben, H.W. Horch et al., submitted for publication), and files listing the BLAST hits of assembly products.
The ‘Search’ link in the top navigation menu takes the user to the main search page of ASGARD, with an introduction to the four search methods provided: (i) Gene Prediction; (ii) Read Name; (iii) GO Annotation and (iv) BLAST Annotation (Figure 3A). The search method introduction text can be shown or hidden using a button on the top left. Users are provided with a drop-down menu containing search options and a second drop-down menu where they can choose the transcriptome they wish to query.

ASGARD Gene Prediction search. The search page gives users access to the Gene Prediction, Read Name, GO Annotation and BLAST Annotation search functions. (A) The input user interface allows users to choose an organism of interest and enter query terms based on D. melanogaster gene names. While entering the search term, users are assisted by an auto-completed list of suggested matching gene names. In this example, a user searching for G. bimaculatus orthologs of Janus kinase (JAK) has begun to enter the name of the D. melanogaster JAK ortholog ‘hopscotch’, which obtained an exact match in the auto-completion list (arrow). (B) The output of the gene prediction search shows predicted ortholog matches, a description of the D. melanogaster gene, schematic representations of and links to matching assembly products.
The ‘BLAST’ link in the top navigation menu takes users to a page where they can search through transcriptome sequences using the NCBI BLAST algorithm (96). Users can query individual transcriptomes from a given organism, or all transcriptome sequences in a single search.
User interface and query implementation
The following sections describe the input and output user interfaces for the five search capabilities of ASGARD.
Search by Gene Prediction
It can be difficult to distinguish the most likely ortholog to a query sequence among multiple results with low E-values obtained by BLAST searches against nr. For ASGARD users wishing to identify putative O. fasciatus, P. hawaiensis and G. bimaculatus orthologs of genes of interest, the most direct route is therefore to use a Gene Prediction search (Figure 3A). Users can choose an organism of interest from the drop-down menu and enter query terms into the search box. Query terms may be a complete or abbreviated D. melanogaster gene name. A link to FlyBase (12) is provided to help users find D. melanogaster gene names. As the user enters a query term, the auto-complete function suggests results ranked in order of best match, retrieved from the pre-computed reciprocal BLAST data to the D. melanogaster proteome (Figure 3A). Only genes with predicted orthologs in the selected transcriptome appear as results of the auto-complete function, allowing users to quickly detect whether a putative ortholog to their gene of interest is available in ASGARD (Figure 3A, arrow). Users can choose a term from the auto-complete list or click the search button. If any predicted orthologs of the query gene are present in the transcriptomes, ASGARD directs the users to a dynamically generated Common Gateway interface (CGI) page that includes a link to each read annotated as a putative ortholog (Figure 3B). The results page also contains the name of the predicted D. melanogaster ortholog, the FlyBase description of the gene and an explanation of the search results display.
Because the current ASGARD transcriptomes were created with tissue-specific samples and were not all sequenced to saturation (21, 22, V. Zeng, B. Ewen Campben, H.W. Horch et al., submitted for publication), many predicted transcripts are incompletely covered with fragmented (non-overlapping) assembly products. As a result, users may obtain multiple assembly products as matches to a query. In addition, multiple splicing isoforms of many genes may be present in the transcriptomes. The results page therefore includes a graphical interface, designed to help users visualize all of the matching different assembly products identified as putative orthologs of the query gene (Figure 3B), and to understand which portions of their gene of interest have sequence coverage in the transcriptomes. In this schematic, a black bar representing the full-length D. melanogaster protein is used as a parent track and beneath it, the matching regions of each assembly product are displayed on individual tracks as grey bars. Tracks of the same length shown in different shades of grey indicate potential isoforms. The unique identifier (read name) of each assembly product result is listed to the left of the schematic and links to all annotation information about each sequence. On this and all other search result pages, explanatory text and result components can be shown or hidden using buttons at the left.
Search by Read Name
The read name search method provides comprehensive annotation information about each transcript. As read names are the unique identifiers of isotigs and singletons, users are unlikely to know these read names a priori, and will therefore perform this search most easily by clicking on read name links returned as results of gene predictor, BLAST annotation, GO annotation or NCBI BLAST searches in ASGARD. Links to or searches for read names direct users to a dynamically generated CGI page containing all annotation data for the specified read name.
The page displays eight types of information for each assembly product (Figure 4): (i) the read name (‘sequence ID’); (ii) the name of the predicted D. melanogaster ortholog obtained by Gene Predictor if applicable; (iii) the GO annotation prediction if applicable, including a link to the GO accession number that allows users to access all transcripts from the query organism with the same GO annotation; (iv) the nucleotide sequence of the predicted coding strand [negative (NEG) or positive (POS) strand is indicated] based on EST Scan results, including a link to the sequence in FASTA format (predicted non-coding sequences are indicated as such above the nucleotide sequence); (v) the predicted amino acid sequence of the assembly product based on EST Scan results if applicable, including a link to the sequence in FASTA format (for predicted non-coding sequences no translation is shown); (vi) a schematic of the predicted protein coding region (white bar) relative to the entire nucleotide sequence of the assembly product (black bar), the lengths of both sequences are indicated; (vii) a list of predicted functional protein domains based on InterPro Scan analysis if applicable, with a schematic representation of the portion of the transcript sequence containing each domain (yellow bars), for each predicted protein domain, links are provided to the relevant protein database where users can obtain further information about each domain and (viii) the pre-computed results of a BLAST search against the NCBI nr database using the assembly product as a query (E-value cutoff 1e−5). BLAST results are listed in order of increasing E-value, and display the species identity and sequence name of the result, E-value, NCBI accession number and a schematic showing the overlapping region of query (white) and subject (black) sequences. The NCBI accession numbers are links to the corresponding GenBank accession.
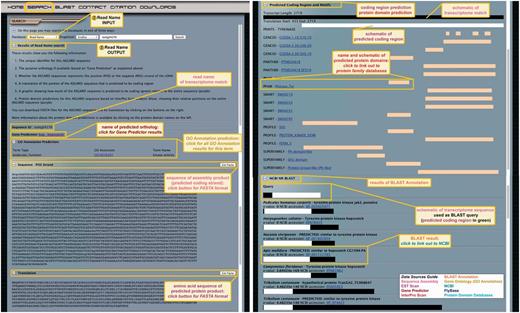
ASGARD Read Name search. The input user interface allows users to enter an assembly product (isotig or singleton) unique identifier (read name) obtained with any of the other search methods (Figures 3 and 5–7). In this example, read name isotig04276 was obtained in a Gene Prediction search (Figure 3) as a putative G. bimaculatus JAK ortholog. The output of read name searches provides all annotation data for the selected assembly product.
In addition to providing complete information on reads retrieved via other searches, the read name search method will also be useful for researchers to revisit a particular sequence of interest for which they have noted the unique ASGARD read name.
Search by GO Annotation
ASGARD users who would like to identify all transcriptome sequences from a given organism that fall into a given GO category can use the GO Annotation search function. Users select an organism of interest from the drop-down menu and enter query terms into the search box (Figure 5A). All or part of GO term may be used as a query and a link to the GO (77) web page is provided to help users find GO terms if necessary. As the user enters a query term, the auto-complete function suggests GO terms that the user may choose to search, or they can simply enter their desired text and click the search button (Figure 5A).
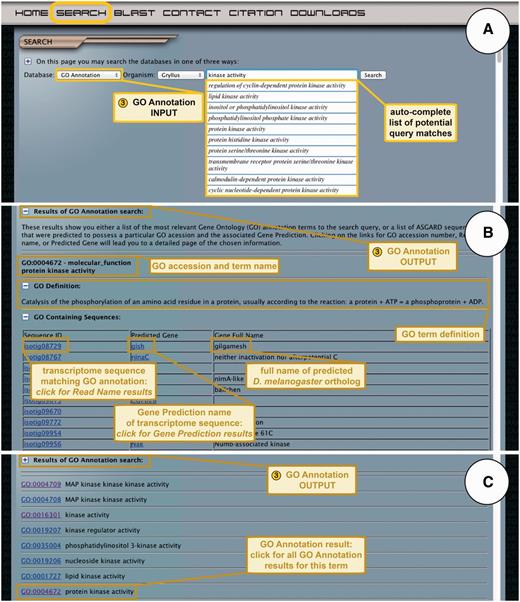
ASGARD GO Annotation search. (A) The input user interface allows users to choose an organism of interest and enter GO term queries, where they are assisted by an auto-completed list of suggested matching GO terms. In this example, a user searching for G. bimaculatus protein kinases enters the query ‘kinase activity’ into the search box. (B) The output of GO annotation searches based on user-defined queries yields a list of transcriptome sequences that map to the chosen GO term, with links to the read name searches for each sequence. This example shows results obtained by using ‘kinase activity’ as a query. (C) The output of GO annotation searches based on selection of auto-completion menu items yields a list of matching GO terms assigned to transcriptome sequences from the chosen organism, with links to all transcriptome sequences assigned to a given GO term. The example shows results obtained by selecting the GO term ‘protein kinase activity’ from the drop-down auto-completion suggestions shown in (A).
In response to user queries, ASGARD utilizes two different search algorithms. The first algorithm is initiated if the user enters a search term and clicks the search button. This algorithm takes the user to a dynamic CGI page with a list of matching GO terms assigned to transcriptome sequences, listed in order of descending relevance (Figure 5B) (relevance is defined as the number of words matching the query possessed by each GO term). GO accession numbers in the results list are links to a page listing transcriptome sequences from their chosen organism that were assigned to the selected GO term.
The second algorithm is used if the user selects a term from the auto-completion list. The auto-completion module uses an exact match algorithm, which means the suggested GO terms must possess each of the search words to be counted as a match. This module shows the first 10 GO terms found under these criteria. Choosing a GO term in this way takes the user to a dynamic CGI page listing transcriptome sequences from their chosen organism that were assigned the selected GO term (Figure 5C). The unique identifiers of these results provide links to the results of the read name search.
Search by BLAST Annotation
Searching for an ASGARD sequence similar to a gene of interest via BLAST is limited by the query sequence, and the Gene Predictor search method is similarly limited by the D. melanogaster proteome sequences. The BLAST annotation search therefore provides users with an alternative method to identify genes that may most closely resemble known sequences from organisms other than D. melanogaster, or that may have been lost in the lineage leading to D. melanogaster. Users select a transcriptome of interest from the drop-down menu and enter a query term, which may be gene name of any organism (Figure 6, top). This prompts a text search through descriptions of all pre-computed BLAST hits against nr for each assembly product in the selected ASGARD transcriptome. The results of the search are displayed on a dynamic CGI page and include the search term and list of BLAST hits in order of increasing E-value (Figure 6, bottom). The results are displayed as described above for the BLAST hit results of the read name search.
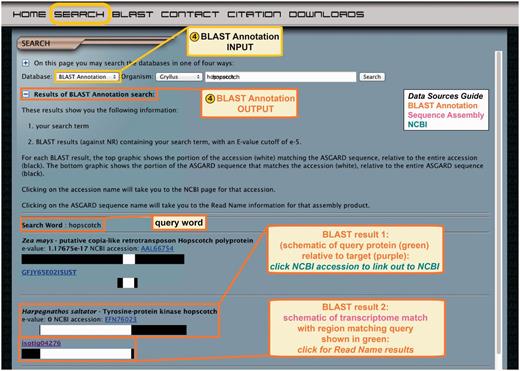
ASGARD BLAST Annotation search. Top: the input user interface allows users to select an organism of interest and enter queries based on gene names from any organism. This example shows what a user might enter to search for G. bimaculatus transcripts similar to JAK orthologs from any organism in nr. Bottom: the output of this search lists BLAST hits against nr whose text descriptions contain the search term, with links to the NCBI accession for each hit, schematic representations of matching transcriptome sequences and links to read name data.
BLAST against NCBI nr sequences
Finally, ASGARD users may search all transcriptome sequences based on nucleotide or protein similarity to nr sequences using the embedded NCBI BLAST module. All ASGARD sequences have been formatted as nucleotide BLAST databases. The user interface mimics that of the NCBI BLAST interface, which is likely familiar to prospective ASGARD users. The transcriptome databases can be queried with a nucleotide sequence using BLASTn or tBLASTx or with a protein sequence using tBLASTn (Figure 7A). The output of these searches is formatted identically to BLAST results obtained through NCBI (Figure 7B). Based on the unique identifier of each sequence in ASGARD, BLAST results are parsed to create a link for each hit that directs the user to the read name results. The bit score of each hit links to the alignment of the query and subject sequences for a given BLAST hit.
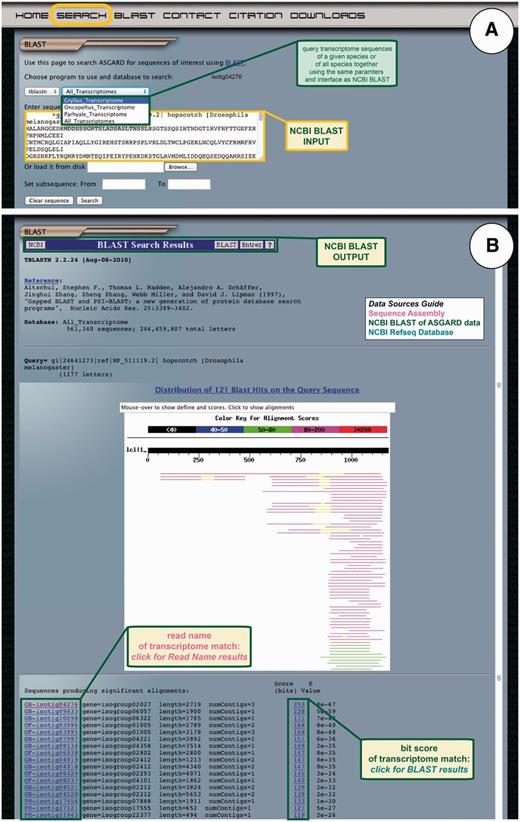
ASGARD NCBI BLAST search. The BLAST page gives users access to the embedded NCBI BLAST function to query transcriptome sequences with a nucleotide or protein sequence of interest. (A) The input user interface allows users to compare the sequence of any gene of interest to the transcriptome sequences from one or all ASGARD organisms using BLAST. In this example, a user chooses the tBLASTn algorithm to search for G. bimaculatus sequences similar to the D. melanogaster JAK ortholog ‘hopscotch’ by using NCBI accession NP_511119 as a query. (B) The output of this search is transcriptome sequences formatted as for the NCBI BLAST algorithm (97). For each match, the unique identifier links to read name data and the bit score links to the BLAST alignment result. Read names in this output are assigned a prefix identifying the species from which the assembly product derives: GB = Gryllus bimaculatus, OF = Oncopeltus fasciatus, PH = Parhyale hawaiensis. This example shows results of the search for JAK-like G. bimaculatus sequences described in (A).
Conclusions and future perspectives
The ASGARD web interface provides public, searchable access to de novo transcriptomes for three emerging model arthropod species. The original descriptions of these transcriptomes (21, 22, V. Zeng, B. Ewen Campben, H.W. Horch et al., submitted for publication) provided SRA accession numbers (Figure 1) and links to raw data and assembly files but the annotated data for O. fasciatus and P. hawaiensis were initially searchable only by text searches through the FASTA format files. ASGARD provides a solution to this problem, allowing users to obtain comprehensive annotation data for each transcriptome assembly product. In the immediate future, ASGARD will also serve as a repository for the results of RNA-Seq experiments, genome sequencing and other NGS applications on ASGARD organisms. We will augment the existing transcriptomes with such data produced by our group and invite other researchers generating NGS data for O. fasciatus, G. bimaculatus or P. hawaiensis to submit their data to ASGARD for processing via our data analysis pipeline and inclusion in the searchable database. To facilitate this, future versions of ASGARD will contain an upload interface for interested researchers to deposit and annotate their sequence data. As new sequence data are added to ASGARD, the baseline assemblies and annotations will also be updated, providing increasingly comprehensive coverage of the transcriptomes of these arthropods.
If and when future genomic data are generated for these organisms, the ASGARD transcriptomes will provide a useful method of immediately validating genome annotations, as all ASGARD data are currently publicly available. At the moment, to our knowledge, there are no public projects planned for sequencing the genomes of P. hawaiensis or G. bimaculatus. However, sequencing of the O. fasciatus genome (https://www.hgsc.bcm.edu/content/i5k-milkweed-bug) has recently been undertaken by the i5k project (http://arthropodgenomes.org/wiki/i5K), and we plan to ensure that the ASGARD database is fully relational with the O. fasciatus genome data when they become available. At the moment, however, this genome project is in its infancy and no final repository or database structure for the genome data has yet been publicly decided upon.
RNA interference (97) and targeted genome editing techniques (98, 99) have extended the power of functional genetic testing to nearly any arthropod organism in principle (100). The examples of long-standing, highly successful organism-specific databases, including FlyBase (12, 101) and VectorBase (102, 103) illustrate that such databases are instrumental in helping researchers make effective use of functional tools and build sustainable research communities. Albeit at a more modest scale, searchable databases such as ASGARD are required to maximize the potential of NGS data for organisms with limited genomic resources, as they make sequence data publicly available in an easily searchable format. We anticipate that ASGARD will be a useful repository and resource for NGS and genomic data generated for additional non-traditional arthropod models, and welcome deposition of sequence data from researchers working on such organisms.
Funding
The Harvard Stem Cell Institute (Seed Grant number SG-0057-10-00 to C.G.E.); the Ellison Medical Foundation (New Scholar Award number AG-NS-07010-10 to C.G.E.); the National Science Foundation (grant number IOS-0817678 to C.G.E.).
Conflict of interest. None declared.
Acknowledgements
Thanks to James Cuff and the Harvard Faculty of Arts and Sciences Research Computing Group for ASGARD suggestions and discussion, Seth Donoughe for suggestions on figure design and members of the Extavour lab for discussion and ASGARD beta testing.
References
Author notes
Present address: Victor Zeng, Stylux Incorporated, 25 Stickney Road, Atkinson, NH 03811, USA

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}