





Abstract
Osteoarthritis (OA) is the most common form of arthritis in the adult population and is a leading cause of disability. OA-related genetic loci may play an important role in clinical diagnosis and disease progression. With the rapid development of diverse technologies and omics methods, many OA-related public data sets have been accumulated. Here, we retrieved a diverse set of omics experimental results from 159 publications, including genome-wide association study, differentially expressed genes and differential methylation regions, and 2405 classified OA-related gene markers. Meanwhile, based on recent single-cell RNA-seq data from different joints, 5459 cell-type gene markers of joints were collected. The information has been integrated into an online database named OAomics and molecular biomarkers (OAOB). The database (http://ibi.zju.edu.cn/oaobdb/) provides a web server for OA marker genes, omics features and so on. To our knowledge, this is the first database of molecular biomarkers for OA.
Key Points
2405 OA-related marker genes were classified based on GWAS, DEG and DMR analysis from 159 publications involving multi-omics data sets.
5459 cell-type gene markers of joints were collected based on recent single-cell RNA-seq data from different joints.
A comprehensive online database named OAOB (http://ibi.zju.edu.cn/oaobdb/) including 6765 OA-related and/or cell-type marker genes was established.
Introduction
Osteoarthritis (OA) is the most common form of arthritis and a leading cause of disability in adult populations (1). Although it is characterized by fluctuating pain and a reduction in physical function (2, 3), this heterogeneous joint disease often presents as cartilage degeneration, remodeling of the subchondral bone and localized inflammation (4).
Studies related to genetics, genomics and epigenetics have uncovered many novel OA-related risk loci in the past decades, and the main cause of its genetic susceptibility may be changes in the regulation of gene expression (5). Emerging evidence suggests that OA genetic risk loci play an essential role in the onset or progression of the disease, are related to clinical symptoms and can be used as promising biomarkers. OA-related biomarkers can fulfill different purposes corresponding to the proposed BIPEDS (burden of disease, investigational, prognostic, efficacy of intervention, diagnostic and/or safety biomarkers) classification (6).
With the rapid development of cutting-edge high-throughput sequencing technologies (e.g. next-generation sequencing and single-cell sequencing) and omics methods (e.g. genomics, transcriptomics and methylomics), a wealth of data have been generated from different joint tissues or cell subtypes correlated with OA disease and are publicly available (7). Additionally, many effector genes have been identified based on the analyses of genome-wide association studies (GWASs), differentially expressed genes (DEGs), differential methylation regions (DMRs) and single-cell RNA sequencing (scRNA-seq). Considering that the huge number of OA-related markers are scattered in the different published literature, there is an urgent need to organize and categorize them, so they can be conveniently and practically used by the OA community.
Despite the importance of categorizing OA-biomarkers, no systematic work has been done on the topic, although a couple of reviews have summarized OA-related biochemical markers (8, 9) with an emphasis on the radiographic or imaginary projects, for example, the Osteoarthritis Initiative (https://www.niams.nih.gov/grants-funding/funded-research/osteoarthritis-initiative).
Overview of different marker genes related to OA
This study retrieved 159 OA-related publications from the Web of Science (Supplemental Table S1) and mined genes associated with OA disease in the literature (Table 1). In total, 2405 genes related to the disease were collected from different joints. These genes are supported by different experimental evidence, with 167 identified based on GWAS (SNVs being associated with 637 genes), 899 DEGs based on bulk RNA-seq data (157 with additional experimental validation) and 1212 supported by DMR analysis (53 being further validated). Moreover, considering the specialty of scRNA-seq data sets, we classified 5459 marker genes identified in specific cell populations in different tissues as ‘cell-type marker’ genes. Of these two groups of marker genes, OA and cell-type related, 1099 are overlapping. These collected genetic biomarkers could be used as predictors in detecting the progression and development of OA. Furthermore, high-confidence effector genes could highlight potential drug intervention targets. They are valuable and better predictive diagnostic biomarkers in measuring early and subtle changes in OA progression compared to the traditional radiographic measures.
Summary of different types of marker genes related to OA
| Evidence sourcesa | Number of marker genes | Key references |
|---|---|---|
| GWAS | 167 | 10–13 |
| DNA methylation | 1212 | 14–17 |
| Bulk RNA-seq/ncRNA-seq | 899/154 | 18–21 |
| Single-cell RNA-seq | 5459 | 22–25 |
| Total | 6,765b |
| Evidence sourcesa | Number of marker genes | Key references |
|---|---|---|
| GWAS | 167 | 10–13 |
| DNA methylation | 1212 | 14–17 |
| Bulk RNA-seq/ncRNA-seq | 899/154 | 18–21 |
| Single-cell RNA-seq | 5459 | 22–25 |
| Total | 6,765b |
Details show the following different sections.
This showed the number of unique genes collected in this study.
Summary of different types of marker genes related to OA
| Evidence sourcesa | Number of marker genes | Key references |
|---|---|---|
| GWAS | 167 | 10–13 |
| DNA methylation | 1212 | 14–17 |
| Bulk RNA-seq/ncRNA-seq | 899/154 | 18–21 |
| Single-cell RNA-seq | 5459 | 22–25 |
| Total | 6,765b |
| Evidence sourcesa | Number of marker genes | Key references |
|---|---|---|
| GWAS | 167 | 10–13 |
| DNA methylation | 1212 | 14–17 |
| Bulk RNA-seq/ncRNA-seq | 899/154 | 18–21 |
| Single-cell RNA-seq | 5459 | 22–25 |
| Total | 6,765b |
Details show the following different sections.
This showed the number of unique genes collected in this study.
Genome-wide meta-analysis based on large-scale multicohort
GWAS has been performed in large-scale OA patient cohorts across various phenotypes. In the early 2010s, based on the comparison between 3177 OA cases and 4894 controls, the 1000 Genomes Project discovered a set of variants associated with OA on chromosome 13 and reported a new OA-related gene, MCF2L (26). Susceptible loci on chromosomes 3 and 7 and the HLA class II/III region of chromosome 6 for knee OA were identified and confirmed from multiple cohorts (27–30).The arcOGEN project studied 7410 selected patients and 11 009 unrelated controls from the UK and identified five genome-wide significant loci on chromosomes 3, 6, 9 and 12, with signals found in different regions close to or within the genes (10). Later, many studies followed and leveraged this UK Biobank and Arthritis Research Osteoarthritis Genetics resources to discover numerous novel risk loci. Some of these loci might cause non-coding, missense variants and are potential new therapeutic targets, such as TFGB1, FGF18, CTSK and IL11 with a supportive evaluation of efficacy in OA (11, 12, 31). With the tremendous and diversified patient population of up to hundreds of thousands of participants from almost every human species (32), a considerable number of risk variants, risk loci and correlative genes were deciphered in the knee, hip, hand, finger, thumb and spine. Also, they were classified in more detail, such as sex-specific and stage-specific (early age-at-onset or late). These results proved OA being a complex, polygenic disease of the whole synovial joints (13, 33, 34).
We analyzed, in detail, the 167 collected markers supported by GWAS evidence and found that they were unevenly distributed on 22 chromosomes, with 1–16 on each chromosome (Figure 1A). In addition, 101 of the 167 markers have other effector or correlative genes, whereas the others influence phenotypes by their variations. Of the total 202 SNP variations on these 167 genes, most variants are annotated on the intron and intergenic regions (Figure 1B). Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment analysis of the 637 effector genes showed that they are closely related to OA, such as in cartilage development or chondrocyte differentiation (Figure 1C, D).
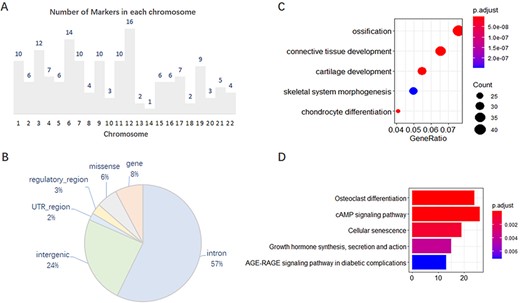
Features of 167 molecular markers supported by GWAS. (A) Number of markers on each chromosome. (B) SNP variants annotated on different genomic regions. (C) GO enrichment of the effector genes. (D) KEGG enrichment of the effector genes.
DNA methylation study in human OA cartilage and blood
DNA methylation profile reflects the tissue identity marked by the epigenetic landscape of cells where the overall methylome differences in the tissue level are commonly reflected in DMRs, whereas cytosine–phosphate–guanine dinucleotides (CpGs) do not harbor this property (35). Recently, genome-wide DNA methylation studies have been performed comprehensively in human articular cartilage. For instance, it has been conducted to reveal epigenetic differences in articular cartilage between Kashin–Beck disease (KBD) and OA (36). Furthermore, it explores epigenomic changes in osteoarthritic subchondral bone and its overlying cartilage (14) and helps understand unique methylomes between normal and OA articular cartilage (16). Also, it distinguishes epigenetic patterns of similar tissues in different body parts (37). Knee and hip cartilage are epigenetically distinct tissues, and their DNA methylation differences have functional properties in regulating the expression of putative joint-specific genes, implicating the approach for future cartilage regeneration (38).
While cartilage and bone for OA are local, blood is systemic. In 2016, blood cells were used to analyze epigenetic premature aging in OA pathogenesis with two DNA methylation age measures (39). The following 5 years have witnessed the growing importance of blood as an effective, easy-to-acquire systemic component in OA research. In 2019, a pilot study investigating the epigenetic pattern in peripheral blood mononuclear cells was conducted to show that quantification of DNA methylation could be adopted as diagnostic or prognostic biomarkers (15). In addition, in the whole blood, HLA-B*27-dependent, independent DNA methylation changes (40), leukocyte LINE-1 hypomethylation and oxidative stress in knee OA were detected (41). In 2021, the distinct DNA methylation patterns of rheumatoid arthritis in peripheral blood T-cells were uncovered (42).
Studies exploring the genomic distribution of CpG sites in groups between OA and normal reveal that the genomic regions, such as gene bodies, exons and regions around transcription start sites (TSSs), may harbor differential methylation sites (43). Also, of the 1212 markers collected from OA-related DNA methylation studies, most genes were differentially methylated in the gene body (76%) and untranslated (UTR) regions (11%), with 34 of them being experimentally verified (Table 2).
Summary of markers from DMR analysis with experimental validation
| Markers from DMR analysis | Methylated region | References |
|---|---|---|
| SOD2; ADAMTS4; MMP-13; COL2A1; H3K9; SOX-5; SOX-6; SOX-9 | Promoter | 44–50 |
| ELF3 | Promoter; TSS200 | 48 and 51 |
| FOXD2; RARA; SOX11 | 1st exon; 3ʹ-UTR | 51 |
| ZHX2; GDF5 | 5ʹ-UTR | 16 and 52 |
| MAFF; ZBTB16; ZNF395; PLEC; ASTN2; CHST11; COG5; DIO2, DIO3; DIO3; DOT1L; FILIP1; FTO; GDF5; GLT8D1; KLHDC5; MCF2L; NCOA3; SUPT3H; TP63 | Body | 16, 17, 53 and 54 |
| Markers from DMR analysis | Methylated region | References |
|---|---|---|
| SOD2; ADAMTS4; MMP-13; COL2A1; H3K9; SOX-5; SOX-6; SOX-9 | Promoter | 44–50 |
| ELF3 | Promoter; TSS200 | 48 and 51 |
| FOXD2; RARA; SOX11 | 1st exon; 3ʹ-UTR | 51 |
| ZHX2; GDF5 | 5ʹ-UTR | 16 and 52 |
| MAFF; ZBTB16; ZNF395; PLEC; ASTN2; CHST11; COG5; DIO2, DIO3; DIO3; DOT1L; FILIP1; FTO; GDF5; GLT8D1; KLHDC5; MCF2L; NCOA3; SUPT3H; TP63 | Body | 16, 17, 53 and 54 |
Summary of markers from DMR analysis with experimental validation
| Markers from DMR analysis | Methylated region | References |
|---|---|---|
| SOD2; ADAMTS4; MMP-13; COL2A1; H3K9; SOX-5; SOX-6; SOX-9 | Promoter | 44–50 |
| ELF3 | Promoter; TSS200 | 48 and 51 |
| FOXD2; RARA; SOX11 | 1st exon; 3ʹ-UTR | 51 |
| ZHX2; GDF5 | 5ʹ-UTR | 16 and 52 |
| MAFF; ZBTB16; ZNF395; PLEC; ASTN2; CHST11; COG5; DIO2, DIO3; DIO3; DOT1L; FILIP1; FTO; GDF5; GLT8D1; KLHDC5; MCF2L; NCOA3; SUPT3H; TP63 | Body | 16, 17, 53 and 54 |
| Markers from DMR analysis | Methylated region | References |
|---|---|---|
| SOD2; ADAMTS4; MMP-13; COL2A1; H3K9; SOX-5; SOX-6; SOX-9 | Promoter | 44–50 |
| ELF3 | Promoter; TSS200 | 48 and 51 |
| FOXD2; RARA; SOX11 | 1st exon; 3ʹ-UTR | 51 |
| ZHX2; GDF5 | 5ʹ-UTR | 16 and 52 |
| MAFF; ZBTB16; ZNF395; PLEC; ASTN2; CHST11; COG5; DIO2, DIO3; DIO3; DOT1L; FILIP1; FTO; GDF5; GLT8D1; KLHDC5; MCF2L; NCOA3; SUPT3H; TP63 | Body | 16, 17, 53 and 54 |
Non-coding RNAs mediated expression in OA progression
Countless studies on the differential expression analysis of target genes relevant to OA provided convincing biomarkers validated in experiments. Starting from the 2010s, expression patterns and novel genes have been intensively studied through transcriptome analysis of major tissues, such as meniscus (55), subchondral bone (56), synovium and fibroblast (18, 57, 58), or in the blood system (59). Genome-wide expression profiles integrated with regulated networks and biological processes would offer comprehensive interpretation and new evaluation of the detected OA candidate genes from multiple aspects. For example, in developing OA, DEG analysis and enrichment results showed that cell cycle-related genes, such as CDK1 and MAD2L1, were downregulated and highlighted in protein–protein interactions (60). Additionally, the novel subnetwork of dysregulated transcription factors identified in bulk RNA-seq data sets of normal and OA knee cartilage tissues was found to be new mediators of abnormal gene expression, representing promising therapeutic targets (19).
Apart from markers collected from bulk RNA-seq data sets, accumulating evidence demonstrated that non-coding RNAs, such as microRNA (miRNA), circular RNA (circRNA), small nucleolar RNA (snoRNA) and long non-coding RNA (lncRNA), could also play an important role in the disease progression by mediating the expression of their target genes (61). For example, the Cyclin D1 gene, CCND1, the target gene of the Wnt pathway validated to have a high expression level in Polynesian and European OA patient groups (62), was modulated by lncRNA FOXD2-AS1 with miR-206 acting as a sponge in chondrocyte proliferation regulation (63). Also, the microRNA-93-5p/CCND1 axis might be activated by SNHG16, and a snoRNA host gene 16 promotes the development of OA by regulating chondrocyte viability (20). A well-known transcription factor in the MMP family named MMP13, reported as a validated differential expressed gene in OA cartilage in multiple data sets (64), was involved in interaction with many non-coding RNAs. In the synovial fluid of patients with knee OA, plasma miR-200c-3p and MMP13 mRNA levels were negatively correlated (65). Also, both arms of miR-675 could affect the expression of MMP13 in the articular chondrocytes (66). Circ_TMBIM6/miR-27a/MMP13 could promote OA-induced chondrocyte extracellular matrix degradation (67). MMP13 and Circ_0136474 could suppress cell proliferation by competitive binding to miR-127-5p (68). With lncRNA-CIR as a sponge for mir-27b, MMP13 expression downregulates miR-27b expression and is associated with IL-1β-induced activation of signal transduction pathways (21, 69).
By reviewing the literature published recently, we found 154 coding genes regulated by 176 non-coding genes, including 160 miRNAs, 11 lncRNAs and 5 circRNAs. While most of them were predicted using bioinformatic tools, a few (Table 3) have been experimentally validated.
Summary of markers regulated by ncRNAs with experimental validation
| Markers | Interactive non-coding RNAs (validated experimentally) | References |
|---|---|---|
| CCND1 | lncRNA FOXD2-AS1, miR-206, miR-93-5p | 20, 62 and 63 |
| MMP13 | miR-27a, miR-27b, miR-127-5p, miR-200c-3p, miR-675, circ_TMBIM6, circ_0136474, lncRNA-CIR | 21 and 64–69 |
| ADAMTS5 | lncRNA-HOTAIR | 70 |
| COL2A1 | miR-455-3p | 71 |
| CTNNB1 | miR-1826 | 65 and 72 |
| MMP1 | miR-675 | 66 and 72 |
| CXCL12 | miR-31 | 73 |
| FOXC1 | miR-138-5p | 74 |
| SMAD3 | miR-203a | 75 |
| Markers | Interactive non-coding RNAs (validated experimentally) | References |
|---|---|---|
| CCND1 | lncRNA FOXD2-AS1, miR-206, miR-93-5p | 20, 62 and 63 |
| MMP13 | miR-27a, miR-27b, miR-127-5p, miR-200c-3p, miR-675, circ_TMBIM6, circ_0136474, lncRNA-CIR | 21 and 64–69 |
| ADAMTS5 | lncRNA-HOTAIR | 70 |
| COL2A1 | miR-455-3p | 71 |
| CTNNB1 | miR-1826 | 65 and 72 |
| MMP1 | miR-675 | 66 and 72 |
| CXCL12 | miR-31 | 73 |
| FOXC1 | miR-138-5p | 74 |
| SMAD3 | miR-203a | 75 |
Summary of markers regulated by ncRNAs with experimental validation
| Markers | Interactive non-coding RNAs (validated experimentally) | References |
|---|---|---|
| CCND1 | lncRNA FOXD2-AS1, miR-206, miR-93-5p | 20, 62 and 63 |
| MMP13 | miR-27a, miR-27b, miR-127-5p, miR-200c-3p, miR-675, circ_TMBIM6, circ_0136474, lncRNA-CIR | 21 and 64–69 |
| ADAMTS5 | lncRNA-HOTAIR | 70 |
| COL2A1 | miR-455-3p | 71 |
| CTNNB1 | miR-1826 | 65 and 72 |
| MMP1 | miR-675 | 66 and 72 |
| CXCL12 | miR-31 | 73 |
| FOXC1 | miR-138-5p | 74 |
| SMAD3 | miR-203a | 75 |
| Markers | Interactive non-coding RNAs (validated experimentally) | References |
|---|---|---|
| CCND1 | lncRNA FOXD2-AS1, miR-206, miR-93-5p | 20, 62 and 63 |
| MMP13 | miR-27a, miR-27b, miR-127-5p, miR-200c-3p, miR-675, circ_TMBIM6, circ_0136474, lncRNA-CIR | 21 and 64–69 |
| ADAMTS5 | lncRNA-HOTAIR | 70 |
| COL2A1 | miR-455-3p | 71 |
| CTNNB1 | miR-1826 | 65 and 72 |
| MMP1 | miR-675 | 66 and 72 |
| CXCL12 | miR-31 | 73 |
| FOXC1 | miR-138-5p | 74 |
| SMAD3 | miR-203a | 75 |
Molecular markers for OA and joints at single-cell resolution
scRNA-seq data analysis allows separating and detecting biomarkers in admixtures of human OA tissues. At single-cell resolution and the whole-transcriptome scale, studies have identified transition pathways, discriminative markers and transcription factors related to specific cell subsets during differentiation in the last 2 or 3 years (22).
Chondrocytes are key cells of cartilage degeneration occurring in diseases, such as OA and KBD, but the heterogeneity of articular cartilage cell types is still unknown (76). Recently, a breakthrough single-cell RNA-seq study revealed the progression of human OA by defining seven chondrocyte populations, including proliferative chondrocyte, prehypertrophic chondrocytes, hypertrophic chondrocytes (HTCs), fibrocartilage chondrocytes (FCs) and three novel populations with distinct functions (77). In another single-cell expression profiling research, HTC, homeostatic chondrocyte and FC were characterized in 480 chondrocyte samples (23). Furthermore, the study compared the transcriptional program and all major cell populations in patient groups between KBD and OA. First, 10 clusters were labeled by cell type according to the expression of previously described markers; second, one novel population with the expression of a new set of markers was identified; finally, the regulatory chondrocyte population was markedly expanded in OA, whereas the homeostatic and mitochondrial chondrocyte populations were in KBD instead (78).
Synovial fibroblasts (SFs) play an important role in OA occurrence and are direct effectors responding to tissue damage and matrix remodeling in synovitis (79). Using the scRNA-seq technique, key genes and pathways on SFs have been revealed (80). Furthermore, the contribution of synovial tissue cell subsets related to joint pain in early and late-stage knee OA has also been identified, exhibiting seven distinct subsets of SFs with differences in their predominance between disease stage and presence of pain. Also, fibroblasts from early to end-stage OA painful synovial sites were found to be associated with differently highly expressive gene sets (81).
Meniscus was especially emphasized on therapeutic consequences and in close relation to developing OA, such as the structural and functional relationship between meniscal tears/extrusion and cartilage loss. Notably, the effect of meniscectomy or meniscal repair was complex (82). The transcriptional dynamics of macrophages from meniscal tissue were analyzed using the scRNA-seq technology in 2020 (83). Results showed characteristic changes in special macrophages in patients with knee OA and healthy individuals by comparing their gene expression profiles. ScRNA-seq was also used to identify cell subsets, their gene signatures in healthy human and degenerated meniscus cells, their differentiation relationships and diversity within specific cell types (84).
Osteoblasts are multifunctional bone cells essential for bone formation, angiogenesis regulation and maintenance of hematopoiesis (85). At the single-cell level, the categorization of primary osteoblast subtypes in vivo in humans has been achieved by performing a systematic cellular taxonomy dissection of freshly isolated human osteoblasts with OA. This helps acquire their gene expression patterns and cell lineage reconstructions (24).
Bone marrow-derived mesenchymal stem cells (BM-MSCs) are multipotent stromal cells that maintain skeletal tissues and differentiate into various mesodermal lineages. scRNA-seq was applied to investigate the transcriptional diversity of BM-MSCs in vivo, showing that they could be codified into distinct subpopulations corresponding to the osteogenic, chondrogenic, adipogenic differentiation trajectories and terminal-stage quiescent cells (86).
Based on the scRNA-seq data sets from different publications, tens of thousands of cell-type marker genes were identified from 10 tissues of 5 joint sites and classified into 48 cell types. We collected 5459 different markers in total (Table 4). Among these cell subtype biomarkers, a part of them (1099) was reported to be OA disease-related markers with variable supporting evidence. For example, 24 of them were confirmed in the GWAS study, 115 in DMR analysis and 181 in bulk RNA-seq or scRNA-seq data. This proves the accuracy and credibility of these cell-type marker genes classified using scRNA-seq.
Summary of 5459 cell-type marker genes for different joint sites collected in this study
| Site | Tissue | Cell types | Marker genes | Key references |
|---|---|---|---|---|
| Knee | Chondrocytes | 11 | 1460 | 76, 81 and 84 |
| Synoviocytes | 8 | 3502 | ||
| HLA-DRA+ synoviocytes | 5 | 1656 | ||
| Meniscus | 10 | 47 | ||
| Meniscus (-synovium) | 3 | 30 | ||
| Synovial | 1 | 809 | ||
| Hip | Osteoblast | 4 | 2589 | 24 |
| Femoral shafts | BM | 3 | 1340 | 86 |
| Human induced-pluripotent stem cells | Chondroprogenitor cells | 3 | 12 | 87 |
| Total | 5,459a |
| Site | Tissue | Cell types | Marker genes | Key references |
|---|---|---|---|---|
| Knee | Chondrocytes | 11 | 1460 | 76, 81 and 84 |
| Synoviocytes | 8 | 3502 | ||
| HLA-DRA+ synoviocytes | 5 | 1656 | ||
| Meniscus | 10 | 47 | ||
| Meniscus (-synovium) | 3 | 30 | ||
| Synovial | 1 | 809 | ||
| Hip | Osteoblast | 4 | 2589 | 24 |
| Femoral shafts | BM | 3 | 1340 | 86 |
| Human induced-pluripotent stem cells | Chondroprogenitor cells | 3 | 12 | 87 |
| Total | 5,459a |
1,099 genes were collected as OA disease-related markers to Table 5.
Summary of 5459 cell-type marker genes for different joint sites collected in this study
| Site | Tissue | Cell types | Marker genes | Key references |
|---|---|---|---|---|
| Knee | Chondrocytes | 11 | 1460 | 76, 81 and 84 |
| Synoviocytes | 8 | 3502 | ||
| HLA-DRA+ synoviocytes | 5 | 1656 | ||
| Meniscus | 10 | 47 | ||
| Meniscus (-synovium) | 3 | 30 | ||
| Synovial | 1 | 809 | ||
| Hip | Osteoblast | 4 | 2589 | 24 |
| Femoral shafts | BM | 3 | 1340 | 86 |
| Human induced-pluripotent stem cells | Chondroprogenitor cells | 3 | 12 | 87 |
| Total | 5,459a |
| Site | Tissue | Cell types | Marker genes | Key references |
|---|---|---|---|---|
| Knee | Chondrocytes | 11 | 1460 | 76, 81 and 84 |
| Synoviocytes | 8 | 3502 | ||
| HLA-DRA+ synoviocytes | 5 | 1656 | ||
| Meniscus | 10 | 47 | ||
| Meniscus (-synovium) | 3 | 30 | ||
| Synovial | 1 | 809 | ||
| Hip | Osteoblast | 4 | 2589 | 24 |
| Femoral shafts | BM | 3 | 1340 | 86 |
| Human induced-pluripotent stem cells | Chondroprogenitor cells | 3 | 12 | 87 |
| Total | 5,459a |
1,099 genes were collected as OA disease-related markers to Table 5.
Development of OAOB database, a database of OA omics and molecular biomarkers
Based on the overall review of our collected marker genes, including 2405 OA disease-related (Table 5), we concluded that these genetic markers have origins from thorough patient cohorts like male and female or the old and child. Furthermore, these genetic markers were from different sites for disease progression, from knee to finger, specifically representing different disease stages. Notably, evidence from different sequencing technologies and experimental methods supported these genetic markers, and nearly a hundred of the genes were supported by evidence from more than two experimental methods. Therefore, these marker genes collected in our study were representative, thoughtful and convincing. The above marker genes are predictive of OA progression and valuable diagnosis biomarkers with superior ability to measure early and subtle changes in OA progression compared to traditional radiographic measures. Additionally, 30 OA marker genes can be targeted by available drugs listed in the DrugBank database (13), which provides another useful clinical approach to treat OA.
Summary of 2405 OA molecular biomarkers (disease-related) in the current release (Release #1) of OAOB database
| Genetic analysis | Expression basedc | ||||||
|---|---|---|---|---|---|---|---|
| OA site or itemsa | GWASb | Bulk RNA | Non-coding RNAe | Single-cell RNA | DNA methylation based | Number of drug targetsa | Total |
| All | 41 | 41 | |||||
| Knee | 39 | 822 (128)d | 141 (46) | 188 | 1136 (37) | 22 | 2216 |
| Hip | 72 | 134 (16) | 9 (9) | 8 | 1000 (32) | 18 | 1265 |
| KneeHip | 41 | 1 | 41 | ||||
| Hand | 10 | 1 | 10 | ||||
| Finger | 5 | 15 | 1 | 20 | |||
| Thumb | 4 | 1 | 4 | ||||
| TMJ | 1 (1) | 1 | |||||
| Sex-specific | 1 | 1 | |||||
| Early-onset all | 1 | 1 | |||||
| Blood | 86 (20) | 26 | 112 | ||||
| Total | 167 | 899 (157) | 154 (65) | 251 | 1212 (53) | 30 | 2405 |
| Genetic analysis | Expression basedc | ||||||
|---|---|---|---|---|---|---|---|
| OA site or itemsa | GWASb | Bulk RNA | Non-coding RNAe | Single-cell RNA | DNA methylation based | Number of drug targetsa | Total |
| All | 41 | 41 | |||||
| Knee | 39 | 822 (128)d | 141 (46) | 188 | 1136 (37) | 22 | 2216 |
| Hip | 72 | 134 (16) | 9 (9) | 8 | 1000 (32) | 18 | 1265 |
| KneeHip | 41 | 1 | 41 | ||||
| Hand | 10 | 1 | 10 | ||||
| Finger | 5 | 15 | 1 | 20 | |||
| Thumb | 4 | 1 | 4 | ||||
| TMJ | 1 (1) | 1 | |||||
| Sex-specific | 1 | 1 | |||||
| Early-onset all | 1 | 1 | |||||
| Blood | 86 (20) | 26 | 112 | ||||
| Total | 167 | 899 (157) | 154 (65) | 251 | 1212 (53) | 30 | 2405 |
Classification of sites or items and drug target genes based on genome-wide association study by Boer et al. (13); TMJ: temporomandibular joint; aex-specific: variations specifically in female patients; early-onset all: variations related to early-onset arthritis.
Number of genes listed here represents genes that have SNVs in gene body or regulatory regions, and these SNVs impact on 637 genes.
Based on differential expressed gene analysis of bulk RNA, non-coding RNA and single-cell RNAs.
Numbers in parentheses represents genes that were experimentally validated.
Numbers in this column represent genes were regulated by non-coding RNAs (lncRNA, circRNA or miRNA) based on ncRNA-seq data.
Summary of 2405 OA molecular biomarkers (disease-related) in the current release (Release #1) of OAOB database
| Genetic analysis | Expression basedc | ||||||
|---|---|---|---|---|---|---|---|
| OA site or itemsa | GWASb | Bulk RNA | Non-coding RNAe | Single-cell RNA | DNA methylation based | Number of drug targetsa | Total |
| All | 41 | 41 | |||||
| Knee | 39 | 822 (128)d | 141 (46) | 188 | 1136 (37) | 22 | 2216 |
| Hip | 72 | 134 (16) | 9 (9) | 8 | 1000 (32) | 18 | 1265 |
| KneeHip | 41 | 1 | 41 | ||||
| Hand | 10 | 1 | 10 | ||||
| Finger | 5 | 15 | 1 | 20 | |||
| Thumb | 4 | 1 | 4 | ||||
| TMJ | 1 (1) | 1 | |||||
| Sex-specific | 1 | 1 | |||||
| Early-onset all | 1 | 1 | |||||
| Blood | 86 (20) | 26 | 112 | ||||
| Total | 167 | 899 (157) | 154 (65) | 251 | 1212 (53) | 30 | 2405 |
| Genetic analysis | Expression basedc | ||||||
|---|---|---|---|---|---|---|---|
| OA site or itemsa | GWASb | Bulk RNA | Non-coding RNAe | Single-cell RNA | DNA methylation based | Number of drug targetsa | Total |
| All | 41 | 41 | |||||
| Knee | 39 | 822 (128)d | 141 (46) | 188 | 1136 (37) | 22 | 2216 |
| Hip | 72 | 134 (16) | 9 (9) | 8 | 1000 (32) | 18 | 1265 |
| KneeHip | 41 | 1 | 41 | ||||
| Hand | 10 | 1 | 10 | ||||
| Finger | 5 | 15 | 1 | 20 | |||
| Thumb | 4 | 1 | 4 | ||||
| TMJ | 1 (1) | 1 | |||||
| Sex-specific | 1 | 1 | |||||
| Early-onset all | 1 | 1 | |||||
| Blood | 86 (20) | 26 | 112 | ||||
| Total | 167 | 899 (157) | 154 (65) | 251 | 1212 (53) | 30 | 2405 |
Classification of sites or items and drug target genes based on genome-wide association study by Boer et al. (13); TMJ: temporomandibular joint; aex-specific: variations specifically in female patients; early-onset all: variations related to early-onset arthritis.
Number of genes listed here represents genes that have SNVs in gene body or regulatory regions, and these SNVs impact on 637 genes.
Based on differential expressed gene analysis of bulk RNA, non-coding RNA and single-cell RNAs.
Numbers in parentheses represents genes that were experimentally validated.
Numbers in this column represent genes were regulated by non-coding RNAs (lncRNA, circRNA or miRNA) based on ncRNA-seq data.
To comprehensively understand, display and preserve the molecular marker genes related to OA disease progression or corresponding cell types, we developed a user-friendly database named OA omics and molecular biomarkers (OAOB, http://ibi.zju.edu.cn/oaobdb/) (Figure 2A, Supplementary File), which collected a total of 6765 marker genes, including 2405 OA disease-related and 5459 cell-type marker genes, and provided a web server to store their omics features and other characteristics (Figure 2B). The database provides a user-friendly interface for clinical and research purposes, such as search marker genes by using key word (e.g. tissue, disease and gene name) or sequences. A genome browser was provided to show the genomic location of all marker genes, their nearby genes by a zooming window, and all related evidence (Figure 2C). Users can easily browse for marker genes from different experimental sources (e.g. markers from scRNA-seq analysis, Figure 2D) and different levels of experimental validation.
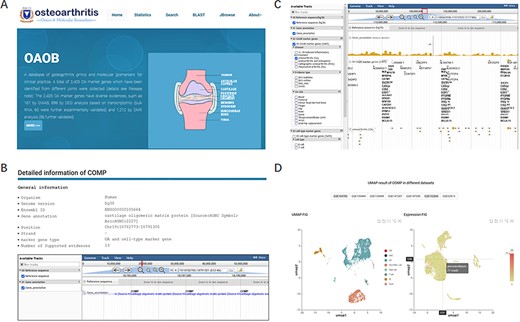
Display of the homepage and function modules of OAOB database. (A) Web homepage of the OAOB database, showing four main functional modules: Statistics, Search, Basic Local Alignment Search Tool (BLAST) and Jbrowse. (B) Using the marker gene cartilage oligomeric matrix protein (COMP) as an example to show all different kinds of information provided. (C) Jbrowse modules of OAOB database. (D) An example showing the marker gene COMP that was supported by scRNA-seq analysis.
We believe that the significant genetic differences of disease severity affected by joint site and sex would help translate the genetic associations into the development of drugs targeting the disease. We wish that the data collected in the database may serve as a solid foundation for future functional and clinical research of OA.
Future perspectives
Integration with a clinical approach to improve treatment
A striking remarkable point is that 30 OA-related marker genes can be targeted by available drugs listed in the DrugBank database (https://go.drugbank.com/), providing attractive insights for connecting the pathogenetic mechanism to the clinical approach treating the disease. With the continuous expansion of information on drugs and targeted sites, in the future, we will update the database by integrating the new information.
Emphasis on the genetic markers from the blood system
In the blood samples of OA patients, quantities of marker genes with clinical significance and experimentally validated were identified by examining the DNA methylation level or the different expression levels relative to normal (Table 5). The results suggest that blood-derived markers may be useful for their predictivity and diagnosis of OA progression, offering advantages over traditional biochemical biomarkers in clinical screening and diagnosis and improving the early care of patients with OA.
Future application of single-cell RNA-seq technology in OA
The highlighted ability of single-cell RNA-seq technology to cluster and group cell populations according to high variable marker gene expression proves to be the key contribution in deciphering cell differentiation and estimating disease development. Additionally, the highly expressive marker genes are good candidates for the identification of the close relationships between cell types and clinical outcomes. Therefore, the scRNA-seq analysis could bring new possibilities in developing diagnostic and therapeutic strategies for OA.
Supplementary data
Supplementary data are available at Database Online.
Funding
National Natural Science Foundation of China (82102646).
Conflict of interest
None declared.
Contributors
L.F., J.L., X.Y. and Q.C. substantially contributed to the conception or design of the work or the acquisition, analysis or interpretation of data for the work. X.Y., X.X. and Y.D. collected the data, and L.X. and Q.C. developed the database. L.F., Q.C. and M.P.T. contributed to drafting the manuscript.
References
Author notes
contributed equally.

{kind=link}
{kind=link}