





Abstract
Synthetic lethality has been widely concerned because of its potential role in cancer treatment, which can be harnessed to selectively kill cancer cells via identifying inactive genes in a specific cancer type and further targeting the corresponding synthetic lethal partners. Herein, to obtain cancer-specific synthetic lethal interactions, we aimed to predict genetic interactions via a pan-cancer analysis from multiple molecular levels using random forest and then develop a user-friendly database. First, based on collected public gene pairs with synthetic lethal interactions, candidate gene pairs were analyzed via integrating multi-omics data, mainly including DNA mutation, copy number variation, methylation and mRNA expression data. Then, integrated features were used to predict cancer-specific synthetic lethal interactions using random forest. Finally, SLOAD (http://www.tmliang.cn/SLOAD) was constructed via integrating these findings, which was a user-friendly database for data searching, browsing, downloading and analyzing. These results can provide candidate cancer-specific synthetic lethal interactions, which will contribute to drug designing in cancer treatment that can promote therapy strategies based on the principle of synthetic lethality.
Database URLhttp://www.tmliang.cn/SLOAD/
Integrated features from multi-omics data were firstly collected.
Cancer-specific synthetic lethal interactions were then predicted using random forest.
SLOAD, a user-friendly database, was constructed based on predicted results.
Introduction
Accumulating evidence has revealed that synthetic lethality (SL) has been an emerging and important therapeutic strategy for cancer treatment by exploiting potential interactions between driving mutations and specific drug targets. A synthetic lethal interaction occurs when the perturbation of two nonessential genes with SL is lethal (1), which can be harnessed to selectively treat cancer via characterizing inactive genes and then targeting the corresponding synthetic lethal partners (2). These genetic interactions between tumor suppressor genes and other genes can simultaneously disrupt both gene functions, which further cause rapid and selective cell death (3). The strategy of SL provides an opportunity for mutations caused by a loss of function without available targeted therapies. The concept of ‘synthetic lethality’ has been widely concerned in cancer treatment, and treatment of cancer-deficient tumors with PARP inhibitors always selectively kills the cancer cells in some cancers (4–9). Thus, this phenomenon caused by synthetic lethal interactions provides a possibility to develop selective anticancer drugs by targeting a gene whose partner is inactive only in tumor cells (1, 10).
Based on the potential application of SL in cancer treatment, it is quite crucial to obtain a data set containing synthetic lethal interactions with higher confidence that can provide data references for precision cancer therapy. In recent years, many studies attempted to find synthetic lethal interactions, especially potential interactions in some model organisms (11–14) and humans (2, 15–21). According to phylogenetic conservation, some human synthetic lethal interactions have been computationally inferred from yeast (22) and collected via using metabolic models and evolutionary characteristics of metabolic genes (23–25). These predicted interactions provide many references for human genetic interactions, but SL in humans is far more complex than we initially thought. Currently, many candidate interactions have been reported based on diverse approaches or algorithms (26–29), even via integrating multiple molecular levels, and these studies largely contribute to revealing the synthetic lethal interactions and promoting their potential application in cancer precision treatment. However, it is not sufficient to further understand these interactions, particularly in diverse cancer types. In-depth understanding of cancer-specific synthetic lethal interactions will contribute to their application in cancer treatment, which will provide an important reference for exploring cancer-specific susceptibilities and further treatments.
Herein, toward the realization of the potential application of the concept of SL in cancer treatment, we aimed to predict and analyze candidate cancer-specific interactions through an integrative multi-omics analysis (Figure 1). Firstly, currently reported candidate genetic interactions were collected; an integrative analysis of multi-omics data analysis was performed to screen molecular features, mainly including DNA level [DNA mutation, copy number variation (CNV) and methylation] and mRNA expression level. Secondly, cancer-specific synthetic lethal interactions in 31 cancers were predicted using random forest (RF). Finally, we constructed a user-friendly and open-access database containing cancer-specific gene pairs and primary analysis. Our study may provide cancer-specific data sources and drug targets for further validation and clinical application in precision cancer therapy.
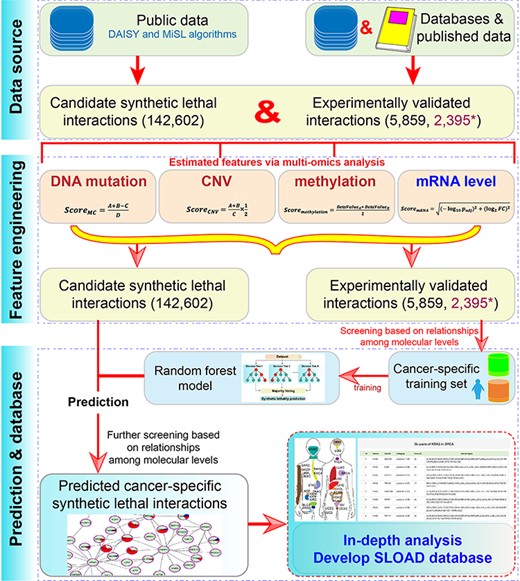
The main flow chart of SLOAD.
Materials and methods
Data resource and data processing
In order to obtain cancer-specific synthetic lethal interactions, candidate genetic interactions were firstly collected to perform further analysis (Figure 1). A total of predicted 142 602 candidate synthetic lethal interactions were mainly obtained from DAISY (19) and MiSL (18) algorithms after removing redundancy. Further, 6518 pairs of experimentally validated synthetic lethal interactions were collected according to published references, including 6033 pairs from Lee et al. (30); 162 pairs validated in Hela, A549 and 293 T cell lines (31); 46 pairs from Syn-lethality database (21); 177 pairs from Srivas et al. (32) and 100 reported gene pairs from published literature (Table S1). Simultaneously, another 2467 pairs without SL in Hela, A549 and 293 T cell lines were also collected (31). Of these, 177 pairs were removed due to contradictory results in different analyses, 554 were removed due to repeated results and a total of 5859 experimentally validated gene pairs and 2395 validated pairs without SL were finally collected.
Multi-omics data, mainly including DNA mutation, CNV, methylation and mRNA expression data in 33 cancer types, were obtained from The Cancer Genome Atlas (TCGA) Web site using the TCGAbiolinks package (33). Genes were removed from further analysis if they were not detected in more than 10n% total samples according to mRNA expression level (reads per kilobase per million mapped reads, RPKM value), mutation, CNV and methylation (beta-value). Of these, n was defined as 7 according to a linear relationship of distributions of the median and average values to ensure that abnormal data had the least effect on the total data distribution. Furthermore, normalized normal data in 27 cancers from Genotype-Tissue Expression (GTEx) database (34, 35) were also downloaded to involve in differential expression analysis (Table S2).
Estimation of features from diverse molecular levels
where A was the sample size of gene A with significant amplification (>1) or significant deletion (<−1), B was the sample size of gene B with significant amplification or deletion and C was the total sample size.
Prediction of cancer-specific synthetic lethal interactions
RF was used to predict cancer-specific synthetic lethal interactions using randomForest R package (38). The cancer-specific training set was firstly constructed based on the correlations of DNA mutation, mRNA expression and CNV levels. Specifically, if one gene was detected mutation, its partner would have a higher expression level and would be amplified more frequently or deleted less frequently harboring the mutation (18, 31). Then, based on the potential relationships of different molecular levels, experimentally validated gene pairs were defined as positive and negative samples. Moreover, some cancers were found with sample imbalance that might influence effective prediction, and a negative sampling strategy was used to improve recall (more than 70% after treatment in all cancer types). The model performance was estimated using a 10-fold cross-validation method, and the model was also evaluated using accuracy, recall and a receive operating characteristic curve to calculate the area under the curve (AUC). Then, for each cancer type, the whole data set was used to construct the classifier. The primary predicted cancer-specific synthetic lethal interactions were collected by classifying the candidate interactions, and further filtering was performed based on strategies mentioned above to ensure the correctness.
Finally, according to predicted cancer-specific results, a user-friendly SLOAD database for data searching, browsing, downloading and analyzing was constructed.
Results
The overall predicted cancer-specific synthetic lethal interactions
The prediction performances using RF were better than our previous study using decision tree in cancers (Figure 2A). Both the accuracy and AUC values showed a significant difference between the two algorithms, indicating better prediction results using RF method (Figure 2B). Integrated feature of multi-omics data had a higher AUC value than the feature at the single molecular level (Figure 2C), despite different cancer types showing varied results. Of these, DNA mutation had a larger contribution to prediction accuracy, while methylation had less effect on the prediction of SL than other molecular levels. These results indicated that integrative analysis of multi-omics data was the best choice to predict cancer-specific synthetic lethal interactions, which provided more data reference for further discussing the potential molecular mechanisms associated with SL.
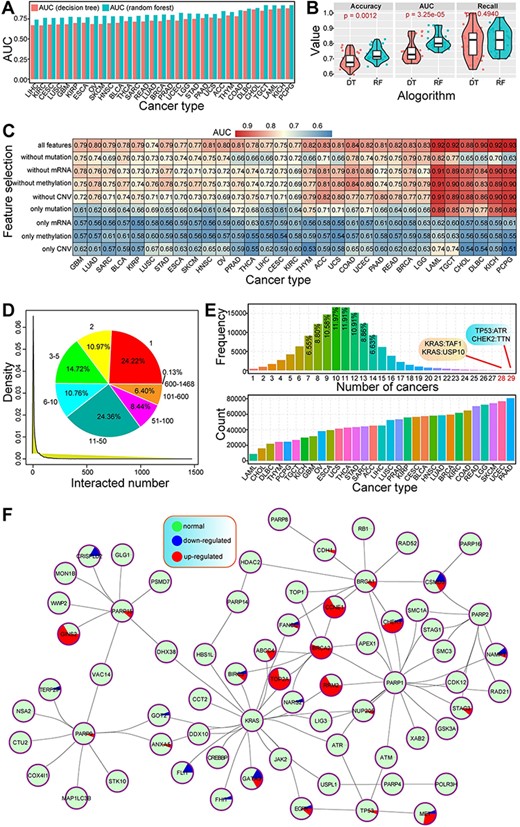
Primary analysis of predicted cancer-specific synthetic lethal interactions.
Herein, a total of 139 035 gene pairs were collected from 31 cancer types, containing 10 377 genes. Of these, most genes (75.78%) were detected in more than one interaction with other genes (Figure 2D). Genes with one interaction were the most popular (24.22%), followed by two interactions (10.97%), indicating that many genes were only found in rare synthetic lethal interactions, while some specific genes may have widespread interactions with other genes. For example, KRAS, PIK3CA and CSMD3 were found more than 1000 interactions. Indeed, KRAS, an oncogenic gene, has been validated by synthetic lethal interaction with other genes, and the determination of synthetic lethal interactions in KRAS oncogene-dependent cancer cells may contribute to revealing novel therapeutic targeting strategies (39–41). These multiple interactions may implicate their important roles in tumorigenesis and further clinical application in cancer treatment based on the concept of SL.
Numbers of predicted gene pairs were different in 31 cancer types, and many synthetic lethal interactions were detected in multiple cancers, especially shared by 7–14 cancers (Figure 2E). Some gene pairs were shared by more than 20 cancers, such as TP53:ATR and CHEK2:TTN were detected in 29 cancer types. The genetic interactions among genes were complex (Figure 2F), especially for those pairs shared by multiple cancers. Some genes were involved in dynamic expression patterns across cancers, implicating the potential biological roles associated with specific cancers. These cancer-specific synthetic lethal interactions can provide candidate drug targets for precision treatment.
Web interface
SLOAD (http://www.tmliang.cn/SLOAD) provides a user-friendly web interface that allows users to query the database via multiple modules, mainly including ‘Search’, ‘Analysis’ and ‘Download’ (Figure 3A). Several search/selection boxes are designed to select a specific cancer type or all cancer types. The corresponding cancer-specific synthetic lethal interactions will be presented via inputting the interested gene symbol or paired genes. Each gene pair also indicates the shared number by diverse cancers (Figure 3B). For selected gene pair, further analysis can be presented, mainly including analysis of mutation, methylation, CNV and mRNA expression levels (Figure 3C), and all of these visible results are also obtained on the Analysis page by inputting the interested genes.

Overview of SLOAD database.
Users can comprehensively browse cancer-specific synthetic lethal interactions and corresponding analysis results of different molecular levels. On the ‘Download’ page, users can obtain free access to the main data sets of cancer-specific synthetic lethal interactions in 31 cancers. The ‘Help’ page provides the detailed information about SLOAD. Furthermore, SLOAD welcomes feedback via the email address presented on the ‘Contact us’ page.
Discussion and future directions
As a strategy in cancer therapy with increasing interests and potential applications, SL has been widely concerned in cancer treatment, especially for recent clinical success (8). Given the importance of SL, many studies aim to comprehensively obtain experimentally verified and computationally predicted candidate synthetic lethal interactions that can provide potential targets for anticancer drugs (18–21, 30, 32, 42–45), which can greatly promote the rapid development of cancer treatment based on the theory of SL (46). Although the concept of SL is an attractive therapeutic strategy, only PARPi has entered the clinic. One of the major hurdles is to identify clinically relevant and robust genetic interaction (47), and a critical challenge in personalized medicine is to identify mutation-specific therapies for a specific cancer type. Cancer-specific SL may be used as a promising cancer therapy, such as ATR and CHK1 inhibitors (48). It is urgent to provide cancer-specific synthetic lethal interactions, especially according to features of multi-omics data.
Herein, based on reported candidate synthetic lethal interactions, we aim to further predict cancer-specific SL via integrating analysis of multi-omics data, mainly including DNA mutation, CNV, methylation and mRNA expression levels. Although DNA mutation is the main contributor in the prediction of synthetic lethal interactions, other molecular levels also contribute to obtaining cancer-specific genetic interactions, and integrating analysis of multi-omics data could provide more information and potential correlations among different molecular levels. According to obtained molecular features, cancer-specific gene pairs are predicted using decision tree and RF, and the latter has the better prediction performance. Then, SLOAD database is constructed according to these predicted results using RF, and the primary analysis is also provided in the database. Taken together, SLOAD provides cancer-specific synthetic lethal interactions via integrating multi-omics analysis and prediction and also presents the primary analysis at the different molecular levels. These cancer-specific synthetic lethal interactions may contribute to discovering novel targets for targeted therapy and promoting further application in precision medicine.
In the future, we will continue to update SLOAD as follows: (i) we will perform an integrative analysis containing other molecular levels, especially the widely concerned non-coding RNAs (ncRNAs). As a class of important regulators in RNA regulatory network, diverse ncRNAs, mainly including small negative regulators, microRNAs and their multiple isoforms, long ncRNAs and circular RNAs, have been focused because of the flexible roles during perturbing gene expression, particularly the cross-talks among various RNAs and subsequent biological pathways. The complex interaction of ncRNAs will enrich our understanding of SL. (ii) More algorithms will be used to improve prediction accuracy, and the relevant data are simultaneously updated. (iii) We will update additional online functions according to user feedback to ensure its value as a user-friendly cancer-specific synthetic lethal interactions database. We expect that SLOAD can contribute to research on cancer treatment based on the theory of SL in precision medicine.
Supplementary data
Supplementary data are available at Database Online.
Funding
This work was supported by the National Natural Science Foundation of China (nos. 62171236 and 61771251), the key project of social development in Jiangsu Province (no. BE2022799), the key projects of Natural Science Research in Universities of Jiangsu Province (22KJA180006), the Qinglan Project in Jiangsu Province, Sponsored by NUPTSF (no. NY220041), the Open Research Fund of State Key Laboratory of Bioelectronics, Southeast University (SKLB2022-K03) and the Priority Academic Program Development of Jiangsu Higher Education Institution.
Conflict of interest
None declared.
Data availability
SLOAD (http://www.tmliang.cn/SLOAD/) is freely available to the public without registration or login requirements.
Abbreviation lists of involved cancers in TCGA
ACC, adrenocortical carcinoma; BLCA, bladder urothelial carcinoma; BRCA, breast invasive carcinoma; CESC, cervical squamous cell carcinoma and endocervical adenocarcinoma; CHOL, cholangiocarcinoma; COAD, colon adenocarcinoma; DLBC, lymphoid neoplasm diffuse large B-cell lymphoma; ESCA, esophageal carcinoma; GBM, glioblastoma multiforme; HNSC, head and neck squamous cell carcinoma; KICH, kidney chromophobe; KIRC, kidney renal clear cell carcinoma; KIRP, kidney renal papillary cell carcinoma; LAML, acute myeloid leukemia; LIHC, liver hepatocellular carcinoma; LGG, brain Lower grade glioma; LUAD, lung adenocarcinoma; LUSC, lung squamous cell carcinoma; OV, ovarian serous cystadenocarcinoma; PAAD, pancreatic adenocarcinoma; PCPG, pheochromocytoma and paraganglioma; PRAD, prostate adenocarcinoma; READ, rectum adenocarcinoma; SARC, sarcoma; SKCM, skin cutaneous melanoma; STAD, stomach adenocarcinoma; TGCT, testicular germ cell tumors; THCA, thyroid carcinoma; THYM, thymoma; UCEC, uterine corpus endometrial carcinoma; UCS, uterine carcinosarcoma.
References
Author notes
Li Guo is an associate professor at the School of Geographic and Biologic Information, Nanjing University of Posts and Telecommunications, China. Her research interests include bioinformatics and computational biology.
Yuyang Dou is a master’s student at the School of Geographic and Biologic Information, Nanjing University of Posts and Telecommunications, China. His research interests include bioinformatics and computational biology.
Daoliang Xia is a master’s student at the School of Geographic and Biologic Information, Nanjing University of Posts and Telecommunications, China. His research interests include bioinformatics and computational biology.
Zibo Yin is a master’s student at the School of Geographic and Biologic Information, Nanjing University of Posts and Telecommunications, China. His research interests include bioinformatics and computational biology.
Yangyang Xiang is a master’s student at the School of Geographic and Biologic Information, Nanjing University of Posts and Telecommunications, China. Her research interests include bioinformatics and computational biology.
Lulu Luo is a master’s student at the School of Life Science, Nanjing Normal University, China. Her research interests include cancer molecular biology.
Yuting Zhang is a master’s candidate at the School of Geographic and Biologic Information, Nanjing University of Posts and Telecommunications, China. Her research interests include bioinformatics and computational biology.
Jun Wang is a professor at the School of Geographic and Biologic Information, Nanjing University of Posts and Telecommunications, China. His research interests include bioinformatics and computational biology.
Tingming Liang is an associate professor at the School of Life Science, Nanjing Normal University, China. His research interests include cancer molecular biology and bioinformatics.

{kind=link}
{kind=link}
{kind=link}