





Abstract
Recently, a new reference transcript dataset [Matched Annotation from the NCBI and EMBL-EBI (MANE) select] was released by NCBI and EMBL-EBI to make available a new unified representative transcript for human protein-coding genes. While the main purpose of MANE project is to provide a harmonized gene and transcript information standard, there is no explicit tissue expression information about these MANE select transcripts. In this report, we tried to provide useful expression profiles of MANE select transcripts in various normal human tissues to allow further interrogation of their molecular modulations and functional significance. We obtained the new V9 transcript expression dataset from the Genotype-Tissue Expression (GTEx) web portal. This new GTEx dataset, based on a long-read sequencing platform, affords better assessment of the expression of alternative spliced transcripts. This tissue expression profiles of MANE select transcripts (TEx-MST) database not only provides the basic information of MANE select transcripts but also tissue expression profiles on alternative transcripts in protein-coding genes. Users can initiate the interrogation by gene symbol searches or by browsing the MANE genes with various criteria (such as genome locations or expression rankings). We further utilized the GENCODE biotype feature to identify the top-ranked protein-coding transcripts by choosing the most expressed protein-coding transcripts from GTEx datasets (both V8 and V9 datasets). In summary, there are 18 083 genes matched between MANE and GTEx. Among them, 13 245 MANE select transcripts matched with the top-ranked protein-coding transcripts in GTEx V9 dataset, which underlined the dominate expression of MANE select transcripts. This TEx-MST web bioinformatic database provides a visualized user interface for the normal tissue expression patterns of MANE select transcripts using the newly released GTEx dataset.
Database URL: TEx-MST is available at https://texmst.ibms.sinica.edu.tw/
Introduction
Protein-coding gene annotation remains challenging, even with the thoroughly interrogated human genome (1–4). The comprehension of human protein-coding genes has not been completely deciphered, and it is still evolving with increasing sequencing data (3, 5). Although determining the primary genome sequence is feasible, comprehensive gene annotation is still a crucial and demanding task (6). In eukaryotic genomes, gene annotation is further complicated by the presence of multiple alternatively transcribed mRNA transcripts generated from a single gene locus in many protein-coding genes (7). Besides alternative exon usages, many more alternative complex transcripts would be generated from diverse transcript start and termination sites (8). Therefore, the establishment of standard annotation references on human protein-coding genes would be useful in Next Generation Sequencing (NGS) data analysis and future orthologous gene annotation pipelines. Currently, the National Center for Biotechnology Information (NCBI) RefSeq project and the European Molecular Biology Laboratory (EMBL) GENCODE project are the primary keystones for gene/transcript annotations and are utilized in numerous genome analysis and annotation procedures (9–11).
Because of inconsistencies across bioinformatics databases and the rapid accumulation of numerous NGS transcriptome datasets, a recent joint project between NCBI and EMBL-EBI was conducted to generate a consistent representative mRNA transcript dataset for human protein-coding genes. The Matched Annotation from the NCBI and EMBL-EBI (MANE) project aims to provide matched consistent transcript annotations for human protein-coding genes and defines one well-curated representative MANE select transcript for each human protein-coding gene (12). These MANE select transcripts will serve as default harmonized transcripts for all human protein-coding genes within the RefSeq and Ensembl databases, which will be valuable for gene analysis, clinical reporting, comparative genomics and integrated multi-omic studies. The current MANE release 1.0 contains 19 062 MANE select transcripts for human protein-coding genes and additional 58 MANE-clinical transcripts. We believe that the MANE dataset will be beneficial for future genomic and transcriptomic research as well as for orthologous gene annotations in evolution studies.
Although the MANE project provides unified exon and transcript structure data for human protein-coding genes, the released MANE dataset does not contain expression information. Transcription is required for the generation of mRNA molecules and provides crucial modulations for functional gene regulations in cells and tissues (13). There is only one MANE select transcript defined for each human protein-coding gene. It is curious to understand more about expression profiles of these exclusive MANE select transcripts, especially their differential expression levels in diverse human tissues. We previously utilized the Genotype-Tissue Expression (GTEx) database to visualize tissue expression modulations of alternatively spliced mRNA transcripts of human protein-coding genes (14, 15). Using GTEx transcriptome data, we generated web tools for visually displaying the expression data on top-ranked transcripts of protein-coding genes (16, 17). However, using short-read NGS data to accurately determine the structure of alternatively spliced mRNA transcripts is associated with some issues (18). Therefore, GTEx has recently released a new version of the transcript expression dataset (V9) based on the third-generation long-read sequencing platform (Oxford Nanopore Technologies). This new dataset would provide a better assessment of alternatively spliced transcripts, thereby reducing concerns related to the precision and quantification of earlier short-read datasets (19–21). Here, we aim to provide a unique user-friendly webtool for tissue expression interrogation of MANE select transcripts mainly based on the GTEx V9 dataset.
Methods
MANE and GENCODE datasets
The MANE dataset was retrieved from the NCBI website. The file used in this study was MANE.GRCh38.v1.0.summary.txt. We then extracted the MANE select transcript information using the MANE_status feature column and other annotation features such as gene symbol, gene name, chromosome location, Ensembl gene ID, Ensembl transcript ID, Ensembl protein ID, NCBI gene ID, NCBI RefSeq NM ID and RefSeq NP ID. The dataset contains 19 062 MANE select transcript records for human protein-coding genes and 58 MANE-clinical transcripts. In addition to MANE select transcripts, the additional MANE-clinical transcripts are annotated for reporting clinically significant variants in certain protein-coding genes.
The GTEx project used the GENCODE V26 dataset as the reference annotation for expression analysis. However, the MANE dataset uses the most recent transcript annotation data. For data on mRNA transcripts and gene structures comparison, both the most recent V40 GENCODE dataset and the GENCODE V26 annotation should be obtained. Thus, the retrieved files were gencode.v40.basic.annotation.gff3 and gencode.v26.basic.annotation.gff3 from GENCODE project (22). Because of the revised difference in certain protein-coding genes and transcripts, we used the V26 GENCODE data for processing GTEx expression data, and we also used the V40 GENCODE data for updated MANE transcript information. Ensembl gene ID was used as the primary key feature for integrated comparison in all datasets. The most recent V40 GENCODE dataset contains 19 988 human protein-coding genes and 87 814 protein-coding transcripts. The current MANE project contains only 19 062 human protein-coding genes and MANE select transcripts. Not all human protein-coding genes have been covered by the MANE project. Moreover, the most updated V40 GENCODE lost 4 MANE genes and 29 MANE select transcripts due to mismatched corresponding IDs.
GTEx short-read (V8) and long-read (V9) expression datasets
The Genotype-Tissue Expression Project is an excellent resource for genotypes and gene expression (14); it is supported by the Common Fund of the Office of the Director of the National Institutes of Health as well as by National Cancer Institute (NCI), National Human Genome Research Institute (NHGRI), National Heart, Lung, and Blood Institute (NHLBI), National Institute on Drug Abuse (NIDA), National Institute of Mental Health (NIMH), and National Institute of Neurological Disorders and Strokes (NINDS). All GTEx data retrieved for this study contains no participant data and adheres to the NIH Genomic Data Sharing guideline. In this study, we solely used the processed transcript expression data (16, 17). We obtained both V8 and V9 normalized transcript expression datasets directly from the GTEx Portal. The V8 short-read dataset was based on the Illumina platform, whereas the V9 dataset was based on the Oxford Nanopore Technologies long-read sequencing platform. The data files were GTEx_Analysis_2017-06-05_v8_RSEMv1.3.0_transcript_tpm for the V8 dataset and quantification_gencode_tpm for the V9 dataset. As described on the GTEx website, both datasets use GENCODE V26 as the annotation reference standard. V8 dataset expression quantifications use the RSEM pipeline (23), whereas V9 dataset quantifications use the flair program (24). Notably, the number of tissues and donor samples used varied between the V8 and V9 datasets. The GTEx V8 dataset covered 54 tissue types from 948 donors, whereas the V9 dataset covered only 14 tissue types. The V8 dataset contains 17 382 sequenced samples, and the V9 dataset contains 88 sequenced samples from 56 donors.
APPRIS annotation information
We included APPRIS dataset for protein structure and evolution conservation reference. APPRIS database selects a single coding sequence (CDS) isoform as the principal isoform for each gene based on the protein features, including conservation (25). Principal isoform scores are numbered from 1 to 5, with 1 being the most reliable (26). We retrieved the APPRIS score data directly from the APPRIS website (https://appris.bioinfo.cnio.es/#/downloads). We used the GENCODE40/Ensembl106 Principal Isoformstxt file for our database information application in this study.
Top-ranked protein-coding transcript in GTEx datasets
In order to handle large datasets, we processed the retrieved GTEx data files with Python scripts in order to divide individual transcript expression data according to each tissue subtypes. Each tissue type would have a separate transcript data file. The average expression values for each transcript were tabulated separately in each tissue and then combined the expression data from all tissue types. In summary, the initial number of transcripts was 199 234 records for the V8 dataset and 149 837 records for the V9 dataset. We also classified the transcript types based on the biotype feature of the GENCODE annotation. The biotype feature primarily consist of nonsense-mediated decay, processed transcript, protein-coding, pseudogene, read-through, stop codon read-through, and TR gene. In previous studies (16), we analyzed the expression levels of each transcript of the respective protein-coding gene including non-coding transcripts. Therefore, the average expression data of transcripts from all different tissues were used to rank the expression levels of transcripts, and CDS lengths or transcript lengths were considered when the expression levels were identical. However, not all transcripts are protein-coding transcripts, as indicated by the GENCODE biotype. Therefore, in this study, we further used the biotype feature to determine the top-ranked protein-coding transcript (TRP-Tx) for each protein-coding gene by selecting only the most expressed protein-coding transcripts. We then assigned a top-ranked protein-coding transcript for each gene in both the GTEx V8 and V9 datasets. These top-ranked protein-coding transcripts were used to match and compare with MANE select transcripts.
TEx-MST database construction
The tissue expression profiles of MANE select transcripts (TEx-MST) database was hosted in a web-hosting Docker engine running on an Ubuntu Linux server. The TEx-MST database was implemented by our laboratory mainly using the PHP programming language onto an Apache web server package in conjunction with the MySQL database. A JavaScript D3 package is also implemented in the webpage for interactive graphical display of transcript expression levels. All transcript expression data of protein-coding genes from the GTEx V8 and V9 datasets are stored as flat files and are then loaded into the MySQL database for the TEx-MST web interface. All data on MANE select transcript are freely accessible at https://texmst.ibms.sinica.edu.tw/. In addition, the processed MANE select transcripts list and top-ranked protein-coding transcripts in GTEx V8/V9 are available for download in the TEx-MST website.
A part of the data statistical analysis and graph illustration were performed using the GraphPad Prism software package (version 9). The significance level of P-value was set at 0.05, as done in our previous reports (27, 28).
Results
The release of the MANE dataset is beneficial because manual curation of MANE select transcripts is laborious and challenging. We believe that the MANE select transcript dataset would be beneficial for gene expression studies and functional genomics research. There are various considerations for recognizing the annotated MANE select transcript as the only representative transcript for each protein-coding gene. Only one MANE select transcript is manually curated for each human protein-coding gene, and it should have a pair of identically annotated sequence data and structure data listed in both the RefSeq and Ensembl GENCODE datasets. For most protein-coding genes, transcriptional expression is essential to produce functional protein products. For specific protein-coding genes, tissue-specific expression is required for ensuring specific cellular and tissue functions. It would be noteworthy to learn more about the expression patterns of MANE select transcripts and their modulations in different tissues.
Top-ranked protein-coding transcript in GTEx
We hypothesize that mRNA transcript expression is required for protein-coding gene functions because protein translation machinery uses mRNAs as templates. The expression levels or patterns among alternatively spliced mRNA transcripts play a significant role in functional modulation. Therefore, we previously used the GTEx database to visualize tissue expression modulations of alternatively spliced mRNA transcripts of human protein-coding genes. The GTEx project is an important resource for systematic studies investigating human gene expression modulations across multiple normal tissues (14, 29). A new V9 dataset is now available for interrogating alternative spliced transcripts on the long-read NGS platform. Thus, this GTEx V9 dataset is more suitable for the interrogation of alternatively spliced isoform expressions.
In this study, we focused on the tissue expression data of MANE select transcripts identified using the GTEx V9 dataset. The top-expressed protein-coding transcripts were first identified based on their expression ranking and GENCODE annotation features (such as biotype, transcript length and CDS length). We first matched the MANE gene ID with the GTEx V9 dataset. The GTEx dataset uses the previous GENCODE V26 annotation; thus, we expected that the gene and transcript IDs will vary between the two datasets. For the protein-coding genes investigated, 18 083 matched gene records and 16 398 matched transcript records were found in both datasets. Interestingly, 14 007 MANE select transcripts matched genes have unchanged transcript numbers/IDs since V26 annotation. However, there are constant updates in the exon structures and lengths in the GENCODE database. Thus, there are also some differences between the GENCODE V40 and MANE gene annotations.
In addition to displaying average expression numbers, we tried to present additional tissue expression profiles and features. We previously focused on the expression profiles of all transcript isoforms and then used the GTEx data to rank all expressed transcripts for human protein-coding genes, including both coding and non-coding transcripts. Intriguingly, not all expressed transcripts of protein-coding genes are protein-coding transcripts (16). Herein, as described in the Methods by selecting top-ranked protein-coding transcripts for protein-coding genes, we have identified one top-ranked protein-coding transcript for each of the 19 591 protein-coding genes in the V8 dataset and 18 516 protein-coding genes in the V9 dataset. Comparing these top-ranked protein-coding transcripts assigned between V8 and V9 GTEx datasets, there are 13 542 protein-coding genes having the same TRP-Tx transcripts. Among the 13 542 common TRP-Tx transcripts, more than 80% of them matched with the MANE select transcripts. In the V9 GTEx dataset, there are 4974 protein-coding genes with different TRP-Tx transcript assignments from V8 dataset. Besides the difference in tissue types used and sequencing depth coverages, there have been reports indicating greater dissimilarities in the transcript expression detections among the Illumina and Oxford Nanopore technology platforms (30, 31). It is possible that part of the variation could be due to the long-read and short-read NGS sequencing platforms.
MANE select and TRP-Tx comparison
We then compared the MANE select transcripts with the V9 top-ranked protein-coding transcripts. Due to the older GENCODE annotation (V26) used in the GTEx pipeline, only 18 083 genes could be matched between the MANE and GTEx datasets with respective Ensembl gene IDs. Among them, the transcript data of 1685 genes could not be further matched between the two datasets. Among these 1685 genes, some genes/transcripts are likely not to be expressed in the tissues of V9 dataset, or these genes have updated transcript annotations in newer GENCODE transcript assembly records (relative to the possibly defunct old transcript IDs). In summary, within 18 083 genes having matched gene IDs, we obtained tissue expression data for 16 398 MANE select transcripts using the available GTEx data.
We then examined the expression rankings of the 16 398 MANE select transcripts. As expected, most of the MANE select transcripts were highly expressed transcripts. The MANE select transcripts consisted of 12 375 Rank 1 transcripts, 1944 Rank 2 transcripts, 821 Rank 3 transcripts, 433 Rank 4 transcripts and 248 Rank 5 transcripts. Only 125 MANE select transcripts were ranked beyond the top 10 transcripts (Figure 1). In the case of Rank 2- or lower-expressed MANE select transcripts, we discovered that non-coding transcripts were frequently the top-expressed transcripts in those genes. Thus, these highly expressed non-coding transcripts are usually excluded from our top-expressed protein-coding transcript criteria. This might be applicable in the MANE-selection processes with the consideration of protein-coding transcripts. We provided the options to view different expression-ranked MANE select transcripts in the TEx-MST website by the expression ranking categories.
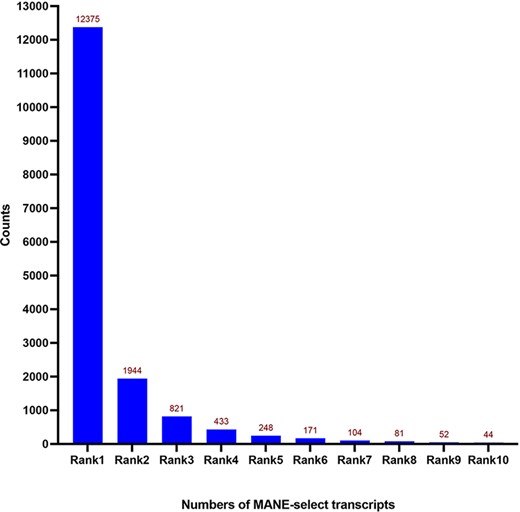
Distribution of expression Ranks (Rank 1–Rank 10) of MANE select transcripts. Most of the MANE select transcripts are the dominantly expressed ones—Rank 1 transcripts according to the GTEx expression dataset. Numbers of genes are shown on top of each column.
When we examined the biotype annotation of the MANE select transcripts (GENCODE V26), in GTEx dataset, 16 MANE select transcripts were listed as non-coding transcripts (processed_transcripts, nonsense-mediated_decay and non_stop_decay). They all have been corrected as protein-coding types in the GENCODE V40 data. In addition to providing expression data, MANE select transcripts are expected to represent full-length translation protein products. We examined the mRNA transcript length and CDS length of MANE select transcripts to determine whether they were also the top-ranked transcripts for protein-coding genes. In the CDS length category, there were 13 851 MANE select transcripts with the longest CDS transcripts (Rank 1 in the CDS-coding ranking). This implies that the designation of MANE select transcripts as those containing the longest translated protein product is optional in some cases. Similarly, in the mRNA transcript length category, only 10 534 MANE select transcripts were the longest; thus, full-length transcripts may not be considered merely based on the length of mRNA molecules due to variations in untranslated region (UTR) regions. These features can be interrogated in the TEx-MST individual gene information pages.
Among matched 16 398 protein-coding genes, we then discovered 13 245 overlapping transcripts between the MANE select transcripts and the V9 top-ranked protein-coding transcripts. Thus, as theorized, most MANE select transcripts are the top-ranked protein-coding transcripts. There are 2219 MANE select transcripts matched only to the V9 dataset, which might be attributed to the difference in long-read sequencing results. However, this result moreover implies that some 3153 MANE select transcripts differed from the V9 top-ranked protein-coding transcripts. There are complex biological evidence considerations in the assignment of MANE select transcripts than our simple expression-based top-ranked transcript collection (12). Another possible reason is the tissue types used in the GTEx V9 long-read sequencing project. Only nine major tissue types were used: adipose, brain (five subtypes), breast, heart (two subtypes), liver, lung, muscle, pancreas and fibroblast. Therefore, some tissue-specific transcripts would not be expressed in these nine tissue types in GTEx V9. When we compared the previous GTEx V8 top-ranked protein-coding transcripts with 54 tissue types, we found additional 1010 MANE select transcripts that matched with the V8 dataset but not with the V9 dataset. This may be partially attributed to the difference in tissue coverage between the two GTEx datasets. These one thousand MANE select transcript genes unique to V8 dataset also enriched in testis, colon, spleen tissues (17, 32), which were not included in the V9 dataset.
For example (Figure 2A and B), the TACR2 (tachykinin receptor 2) gene is highly expressed in the digestive system (the colon, esophagus, stomach and small intestine). The MANE select transcript has the transcript ID of ENST00000373306, which was identified as the top-ranked protein-coding transcript in the V8 dataset. However, in the GTEx V9 dataset (lacking transcripts for the digestive system organs), the other transcript ENST00000373307 was discovered to be our top-expressed protein-coding transcript. Therefore, the TACR2 MANE select transcript was only matched with the V8 top-ranked protein-doing transcript but not in the GTEx V9 data. Therefore, there would be needs for different protein-coding genes, and we provided both the GTEx V8 and V9 expression data for users.
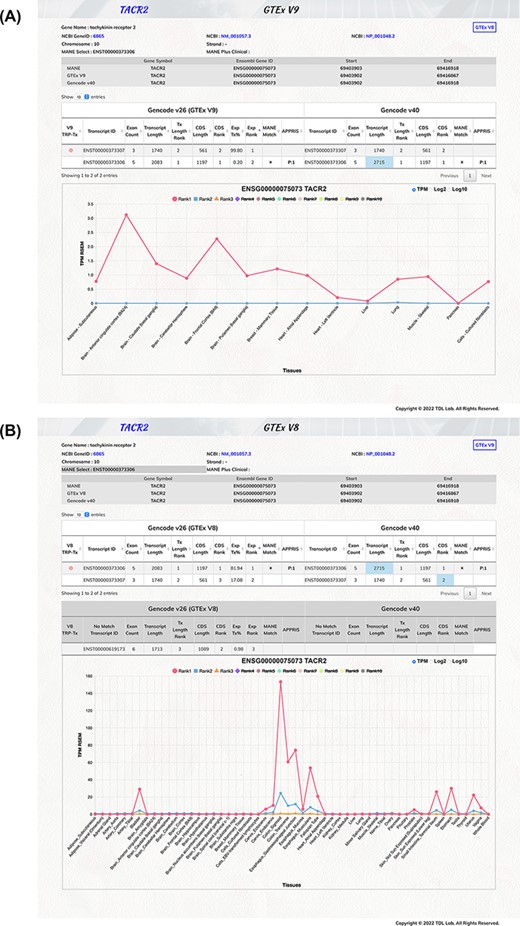
Illustration of TEx-MST gene information webpage for tachykinin receptor 2 (TACR2) protein-coding gene. Basic gene and transcript information of the protein-coding gene are provided. The main transcript expression data table is displayed to provide the GTEx and GENCODE information. The top-ranked protein-coding transcript is marked by a red circle and the MANE select transcript is marked by a star symbol. The important expression graph is provided for Rank 1 to Rank 10 transcripts at the bottom chart of the webpage. (A) GTEx V9 (long-read) expression information is displayed; (B) GTEx V8 (short-read) expression information is displayed. Please note that there are more tissue types in the V8 dataset. The TACR2 gene is mostly expressed in the digestive system.
TEx-MST database
To provide a visualized expression database of the MANE select transcripts, we developed a TEx-MST informatic web tool to visualize tissue expression profiles of human MANE protein-coding genes. Users can examine the tissue expression profiles of MANE select transcripts by using the ‘Gene Symbol’ search function or by their chromosome locations. Some MANE select transcripts exist without their matching GTEx expression data; thus, we categorized the 19 062 MANE gene records into four categories: (i) 13 245 MANE select transcripts with matching GTEx V9 top-ranked protein-coding transcripts; (ii) 3153 MANE select transcripts with different top-ranked GTEx protein-coding transcripts; (iii) there are 1685 genes with matched gene ID, but the transcript ID of MANE select transcripts were not found in the GTEx V9 dataset and (iv) remaining 979 MANE genes without their matching gene IDs with GTEx records (illustrated in Figure 3). In these categories, in addition to listing by their chromosome locations, we listed and indexed the MANE select transcripts by their expression percentage within the protein-coding genes; by their expression rankings and by their expression TPM values. However, for protein-coding genes in (iii) and (iv) categories, there is no GTEx expression data available; therefore, we listed only the GENCODE V40 information in these gene pages.
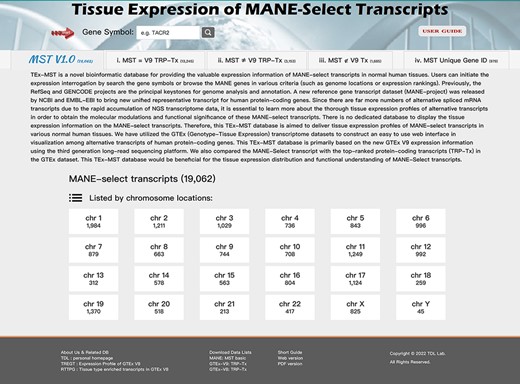
The TEx-MST database web page. We have established a web resource for accessible interrogation on individual MANE select transcripts. There are 19 062 protein-coding gene records in current V1.0 release of MANE project. We further classified them into four categories: (i) matched with GTEx V9 top-ranked protein-coding transcripts—13 245; (ii) not-matched with GTEx V9 top-ranked protein-coding transcripts—3153; (iii) not included in the GTEx V9 transcript list—1685 and (iv) genes not found in the GTEx dataset. A simple user guide is provided for easy access, and users can study the gene of their interests by searching with the gene symbol.
In the individual gene data page (Figure 2), basic gene data is provided at the top, which includes gene name, NCBI gene ID, NCBI RefSeq NM and NP ID, chromosome, strand, MANE select transcript ID and MANE-clinical transcript ID (if any). The start and end chromosome positions (MANE, GTEx and GENCODE) are also listed in a short table. Please note that the MANE annotation might differ from the recent GENCODE V40 annotation. The main transcript data table is then displayed in the middle. In the first column, the top-ranked protein-coding transcripts are marked by a red circle. The MANE select transcript is marked by a star symbol. The GTEx transcript data and the updated GENCODE V40 transcript data are displayed side-by-side for users to learn more about the structure and expression data among all transcripts annotated for this protein-coding gene. The other displayed features include exon numbers, transcript length with its ranking, CDS length with its ranking, MANE select transcript and APPRIS score. Additional transcript data are displayed in a second table for transcripts missing from either GTEx or GENCODE V40. In the bottom panel, the expression graph chart is provided for Rank 1 to Rank 10 transcripts (the top three ranked transcripts are initially displayed and others can be visualized by users). Users can specifically select (on and off) any ranked transcript by clicking the ‘RankX’ symbol in the graph legend. The expression scale can be changed to log2 or log10 scale for better expression inspection of transcripts with lower expression. The default web page is mainly linked to the GTEx V9 expression data, and a link button on the top right corner can be clicked to display extra V8 expression data. As mentioned earlier, the GTEx V9 used 14 tissue types, whereas the GTEx V8 used 45 tissue subtypes. For some genes, it is useful to examine their expression profiles in additional tissue types of V8 dataset. Therefore, in TEx-MST database, users can examine both V8 short-read and V9 long-read expression data freely by their research needs.
We also included APPRIS annotations in this study to examine the functional conservation feature of the MANE select transcripts. APPRIS, created by professor M. L. Tress, is an excellent database that provides reliable data on protein structures and evolutionary cross-species conservation. A single main protein isoform is often generated by most protein-coding genes. The database provides APPRIS annotations for alternatively spliced transcripts for many model organisms and selections of such principal isoforms for protein-coding genes. By including the APPRIS score, 12 771 MANE select transcripts are annotated as Principal 1, 1477 are annotated as Principal 2 and 1075 are annotated as Principal 3. The APPRIS score of Principal 1 implies that this transcript is the only isoform based on the core APPRIS computation modules. There is a good agreement with the APPRIS score and MANE select transcripts. We also discovered that nearly 90% of Principal 1 transcripts matched with our V9 top-ranked protein-coding transcripts. We believe that this APPRIS score feature would be useful for users to learn about investigating the biological functions and conservation of the MANE select transcripts.
Discussion
The NCBI RefSeq dataset previously served as a benchmark annotation standard in bioinformatics and genomic analysis pipelines, including in our previous studies (27, 33–35). With ever-increasing NGS sequencing reads from various transcriptomic studies, additional alternative spliced transcripts have been discovered, generating inconsistent transcript annotations among various bioinformatic databases. Different annotation standards or even various updated versions within the same datasets could lead to data consistency issues. The MANE project is beneficial, as it provides matched consistent transcript annotations for human protein-coding genes. The well-defined MANE select transcripts serve as harmonized transcripts for all human protein-coding genes, which will be valuable for future bioinformatic studies. Well-established gene expression databases are available for alternatively spliced transcripts (36, 37), such as HPA and GTEx, but no database has been developed to visualize MANE select transcript expression data for human tissues. This is the first database for examining MANE select transcript expression in different tissues.
For most protein-coding genes, transcriptional expression is essential to produce functional protein products. Functional modulations of many genes are regulated by their transcriptional activities, especially multiple alternatively spliced mRNA transcripts. Tissue-specific transcript expression is often used for implementing exclusive cellular and tissue functions for certain protein-coding genes, which are crucial regulatory mechanisms in the developmental process (38, 39). Gaining a comprehensive understanding of the expression patterns of MANE select transcripts and their modulations in various tissues would be beneficial for functional genomic studies. As reported previously, many protein-coding genes have a single dominant transcript (40, 41), and it is not surprising that MANE select transcripts are often the dominant transcripts in human protein-coding genes and match with the top-ranked protein-coding transcripts in GTEx dataset discovered here.
The GTEx dataset also provides important tissue expression data for normal tissue types. The GTEx project is a significant resource that provides genome variations and mRNA expression in normal human tissues (42), which can be used for the systematic evaluation of genetic variations and gene expression modulations in multiple tissues (29). Particularly, we used the new V9 long-read expression dataset, which is based on the nanopore (ONT) sequencing platform. Few databases provide the transcript expression data generated from the third-generation NGS technology; these databases often focus on the gene expression profiles of various species. Only a small number of datasets provide the tissue expression patterns of protein-coding genes. Our TEx-MST database is a unique and valuable database to provide long-read NGS data on the alternatively spliced transcript expression data for MANE select transcript. We also provided additional features on the MANE select transcripts, such as their expression rankings, transcript length rankings and CDS length rankings. These details would be beneficial for biomedical researchers to conduct further genomic studies. This database would be useful for understanding tissue expression and conducting functional studies involving standard reference MANE select transcripts.
Conclusion
We used the GTEx V8 and V9 expression dataset to create a web database for visualizing the expression of MANE select transcripts in various human tissue types. This bioinformatic web tool is useful for analyzing the tissue expression patterns of MANE select transcripts. It is also possible to compare the GTEx and updated GENCODE gene/transcript information in MANE select transcripts for better functional analyses on the MANE select transcripts.
Acknowledgements
This work was supported in part by funding from Academia Sinica and from the National Science and Technology Council (109-2311-B-001-013-MY3), Taiwan.
Author contributions
K.-F. Tung retrieved and processed the MANE, GENCODE and GTEx information, as well as constructed the whole TEx-MST website. W.-c. Lin supervised the experiment and prepared the manuscript. All authors reviewed the manuscript.
Conflict of interest.
The authors declare no competing interests.
Data availability
All MANE select transcript tissue expression information can be accessed with no restriction by following link at https://texmst.ibms.sinica.edu.tw/.

{kind=link}
{kind=link}
{kind=link}