





Abstract
BioC is a simple XML format for text, annotations and relations, and was developed to achieve interoperability for biomedical text processing. Following the success of BioC in BioCreative IV, the BioCreative V BioC track addressed a collaborative task to build an assistant system for BioGRID curation. In this paper, we describe the framework of the collaborative BioC task and discuss our findings based on the user survey. This track consisted of eight subtasks including gene/protein/organism named entity recognition, protein–protein/genetic interaction passage identification and annotation visualization. Using BioC as their data-sharing and communication medium, nine teams, world-wide, participated and contributed either new methods or improvements of existing tools to address different subtasks of the BioC track. Results from different teams were shared in BioC and made available to other teams as they addressed different subtasks of the track. In the end, all submitted runs were merged using a machine learning classifier to produce an optimized output. The biocurator assistant system was evaluated by four BioGRID curators in terms of practical usability. The curators’ feedback was overall positive and highlighted the user-friendly design and the convenient gene/protein curation tool based on text mining.
Database URL: http://www.biocreative.org/tasks/biocreative-v/track-1-bioc/
Background
Understanding the organization of molecular interactions is fundamental for comprehending how cellular networks regulate homeostasis and cellular response to external stimuli (1). Thus, many efforts have been made to systematically capture published experimental evidence and make it available for computational approaches through databases such as BioGRID (http://thebiogrid.org) (2), IntAct (3, 4) and DIP (4). In order to facilitate the annotation process a number of text-mining approaches have been attempted with various degrees of success at different stages of the annotation pipeline (5).
The purpose of the BioC (6) track in BioCreative V was to create BioC-compatible modules which complement each other and can be seamlessly integrated into a system that assists BioGRID curators. BioC is a minimalist approach to interoperability for biomedical text mining. It is an XML format to share text data and annotations and comes with a simple library to read/write that data in multiple languages (http://bioc.sourceforge.net). In previous BioCreative workshops, great emphasis was given to the identification of protein–protein interactions (PPI). The PPI track (7–9) was divided into subcategories and each task was addressed independently, i.e. article classification, interaction pair extraction, interaction sentence classification and experimental method identification. The user interactive track (IAT) (10–12) promoted the development of annotation systems that can assist in biocuration tasks by bringing text mining tool developers and database curators together. Nonetheless, probably due to lack of interoperability or sub-optimal performance, no attempt was made to integrate such modules into a single annotation tool.
With this in mind, and considering that the previous BioC track focused on releasing BioC resources such as datasets and biomedical NLP tools, the BioCreative V BioC track was focused on a more practical problem: the collaborative creation of a biocurator assistant tool tailored for BioGRID database curators.
The main goals of the BioC track were:
Define a collaborative task for molecular interaction information extraction, so each team can develop a module independently, but can also use other modules’ outputs.
Develop practical BioC-compatible molecular interaction tools by combining or improving existing methods for full-text articles.
Combine interoperable BioC components to produce a biocurator assistant tool guided by BioGRID curators.
Generate annotated full-text benchmark datasets for the development and the final evaluation of the curation interface.
The BioC track was organized into eight different tasks. Eight teams created the modules that were cooperatively used to identify and annotate molecular interaction information from full-text documents. Using these results, one team implemented a visual interface that was then evaluated by BioGRID curators.
The paper is organized as follows. In the next section, we describe the tasks for the BioCreative V BioC track. This is followed by the description of how interoperability was achieved among teams, participants’ system descriptions, and a description of our machine learning-based merging process for submitted predictions. Finally, user feedback from BioGRID curators is discussed and conclusions are drawn.
Tasks, data and interoperability
One distinctive feature of the BioC track was the focus on collaboration and synergy among participating teams. The organizers promoted a collaborative framework and helped each team to collaborate with the others for building an integrated annotation system. Figure 1 outlines the tasks defined for the BioGRID biocurator assistant tool:
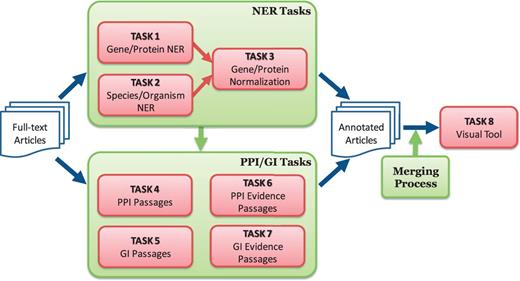
Overview of BioCreative V BioC track. The track consists of named entity recognition (NER), protein–protein interaction (PPI), genetic interaction (GI) and visual tool tasks. The NER tasks include gene/protein NER, species/organism NER and gene/protein normalization. The PPI/GI tasks include finding passages with PPI/GI information (PPI/GI Passages), passages with PPI experimental methods (PPI Evidence Passages) and passages with GI types (GI Evidence Passages).
Task 1, ‘Gene/protein named entity recognition (NER)’—Identification of gene/protein mentions. Participating teams combine results from existing tools or develop their own methods to improve NER performance.
Task 2, ‘Species/organism NER’—Identification and normalization of species/organism names. Participating teams either combine results from existing techniques or propose a new way for identifying species/organisms.
Task 3, ‘Normalization of gene/protein names’—Identification of gene/protein IDs based on gene/protein names and species/organisms mentioned in surrounding text. Previous BioCreative datasets may be used for system development. Systems can optionally use prediction results from Tasks 1 and 2.
Task 4, ‘Passages with PPIs’—Identification of passages describing PPIs, e.g. ‘Aip1p interacts with cofilin to disassemble actin filaments’. Physical interactions may appear in single or several sentences. Participating team(s) may use the PPI corpora (http://corpora.informatik.hu-berlin.de) such as BioCreative, BioNLP Shared Task, AIMed and LLL for training, but they also can develop additional training data.
Task 5, ‘Passages with genetic interactions (GIs)’—Identification of passages reporting GIs, e.g. ‘UBP2 interacts genetically with RSP5, while Rup1 facilitates the tethering of Ubp2 to Rsp5 via a PPPSY motif’. GIs may appear in single or several sentences. The BioGRID set may be used for creating a training set.
Task 6, ‘Passages with experimental methods for physical interactions (PPI evidence passages)’—Identification of passages describing experimental methods used for the discovery of physical interactions, e.g. ‘A two-hybrid-based approach using cofilin and actin mutants identified residues necessary for the interaction of actin, cofilin and Aip1p in an apparent ternary complex’. There are 17 experimental methods defined in BioGRID (Table 1). For this task, BioGRID, MINT and/or IntAct databases may be used for training data.
Table 1.PPI experimental methods and GI interaction types defined in BioGRID
PPI experimental methods GI interaction types Affinity Capture-Luminescence
Affinity Capture-MS
Affinity Capture-RNA
Affinity Capture-Western
Biochemical Activity
Co-crystal Structure
Co-fractionation
Co-localization
Co-purification
Far Western
FRET
PCA
Protein-peptide
Protein-RNA
Proximity Label-MS
Reconstituted Complex
Two-hybrid
Dosage Growth Defect
Dosage Lethality
Dosage Rescue
Negative Genetic
Phenotypic Enhancement
Phenotypic Suppression
Positive Genetic
Synthetic Growth Defect
Synthetic Haploinsufficiency
Synthetic Lethality
Synthetic Rescue
Detailed information can be found in http://wiki.thebiogrid.org/doku.php/experimental_systems.
Task 7, ‘Passages with GI types (GI evidence passages)’—Identification of passages describing GI types, e.g. ‘Synergy of FLAP1 and p300 for enhancement of transcriptional activation by beta-catenin, LEF1/TCF and AR’. These passages may overlap with the ones from Task 5. However, for Task 7, a type of GI should be clearly shown. There are 11 interaction types defined in BioGRID (Table 1). The BioGRID set may be used for training data.
Task 8, ‘Visual tool for displaying various annotations’—Development of a visualization tool for highlighting annotation results from other tasks above. The tool should allow easy navigation and display user-selected annotations. A participating team should work closely with biocurators in BioGRID in order to develop a visualization tool that curators find most useful.
Unlike other BioCreative tasks, no official training/test set was released for the BioC track. This highlighted the nature of this challenge in encouraging its participants to develop practical tools by combining or improving existing resources. Table 2 summarizes the resources that each team utilized for the development and optimization of their methods.
Datasets used and created by participating teams
| Teams | Tasks | Datasets | URL |
|---|---|---|---|
| Matos et al. (T2) | NER (Gene/protein) + normalization | BioCreative II Gene Mention Recognition corpus | http://www.biocreative.org/resources/corpora |
| NER (Species/organism) + normalization | LINNAEUS | http://linnaeus.sourceforge.net/ | |
| Batista-Navarro et al. (T3) | NER (Gene/protein) + normalization | CHEMDNER GPRO | http://www.biocreative.org/resources/corpora/chemdner-patents-gpro-corpus/ |
| NER (Species/organism) + normalization | S800 | http://journals.plos.org/plosone/article?id=10.1371/journal.pone.006539 | |
| Singh et al. (T4) | NER (Gene/protein) + normalization | BioCreative II Gene Mention Recognition corpus | http://www.biocreative.org/resources/corpora |
| NER (Species/organism) + normalization | S800 | http://journals.plos.org/plosone/article?id=10.1371/journal.pone.0065390 | |
| NER (Gene/protein/species) evaluation | IGN corpus | https://sites.google.com/site/hongjiedai/projects/the-ign-corpus | |
| Peng et al. (T6) | PPI passages | 20 in-house full text documents | http://proteininformationresource.org/iprolink/corpora |
| AIMed corpus | ftp://ftp.cs.utexas.edu/pub/mooney/bio-data | ||
| Aydin et al. (T7) | PPI experimental method passages | In-house developed corpus | |
| Kim and Wilbur (T8) | PPI passages | BioCreative PPI corpus | http://www.biocreative.org/resources/corpora |
| Two in-house developed corpora | |||
| Islamaj Dogan et al. (T8) | GI passages | Two in-house developed corpora | http://bioc.sourceforge.net |
Participating teams proposed a method for each task and created new training data or selected from existing training sets. Ten teams submitted task proposals in March, however, one team later withdrew. By the July submission deadline, eight teams had contributed 24 runs addressing Tasks 1–7 and one team built the visualization tool. A submitted run contains predicted text, e.g. gene/protein/organism names or PPI/GI passages, optionally with normalized IDs for gene/protein/organism names. Task 5 was not performed specifically, but it was considered covered by Task 7. Table 3 shows submitted runs for each task for each team. The number of runs varied from 3 to 6 except for the visual interface task.
Submitted runs from nine participating teams
| Team | Task 1 | Task 2 | Task 3 | Task 4 | Task 6 | Task 7 | Task 8 |
|---|---|---|---|---|---|---|---|
| T1 | 1 | ||||||
| T2 | 1 | 1 | 1 | ||||
| T3 | 1 | 1 | 1 | ||||
| T4 | 1 | 1 | 1 | ||||
| T5 | 1 | ||||||
| T6 | 4 | ||||||
| T7 | 2 | ||||||
| T8 | 1 | 2 | 4 | ||||
| T9 | 1 | ||||||
| Total | 4 | 3 | 3 | 6 | 4 | 4 | 1 |
To boost the synergy effect of using multiple runs, we (T8) produced additional results for Tasks 4 and 6. Only one team was selected for Task 8 as it was to develop a user interface.
Achieving interoperability and improving collaboration
One of the goals of the BioC track was to provide a medium where different teams could work independently to produce a more advanced and a more sophisticated system in the given time. This track started with a series of webinars describing the BioC format and the BioC tools, and then continued with regular online conferences where teams shared their progress, questions and suggestions. Figure 2 describes how BioC communication facilitated the sharing of data and annotations produced.
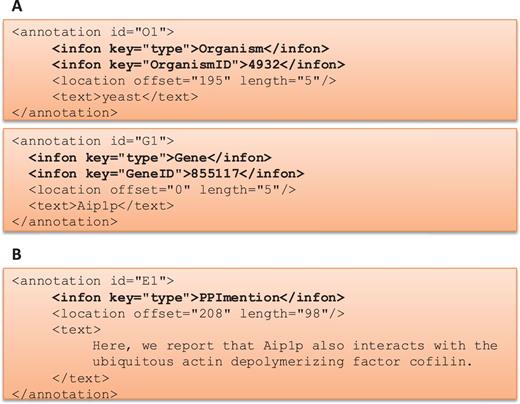
BioC Format for BioCreative V BioC track. (A) BioC format to share annotations for named entity recognition tasks: gene/protein and organism mentions and normalization. OrganismID and GeneID are NCBI Taxonomy ID and Entrez Gene ID, respectively. (B) BioC format to share annotations for the molecular interaction tasks: protein–protein interaction mention and evidence (PPImention, PPIevidence) and genetic interaction mention and evidence (GImention, GIevidence).
Materials and Methods
All teams were invited to provide data processed with their individual systems optimized for the tasks detailed above. Here, we give brief descriptions of the individual systems that contributed to the BioC track.
System descriptions
Namedentityrecognitionsystems
The following teams contributed runs for named entity recognition for Tasks 1, 2 and 3. Neji (13) and Argos (14) were previously equipped with modules to read and write data in BioC format, while the NTTMUNSW system (15), newly developed for this task, created a C# library for reading/writing data in BioC format. Full text articles were processed for gene, protein and species mention recognition and normalization, and the outputs were submitted to the task organizers for availability to the teams working on the other tasks and inclusion in the complete biocurator assistant system.
Neji
Team members: Sérgio Matos, André Santos, David Campos and José Luís Oliveira.
Neji is a biomedical concept recognition framework that uses efficient dictionary-matching, machine learning models, and multi-threaded document processing (13). The gene/protein mention recognition system (Task 1) consisted of a second-order conditional random fields model with orthographic, morphological, dictionary-matching and local context features, as described in (16). This model was trained and tested on the BioCreative II gene mention recognition corpus (17). The species/organism mention recognition system (Task 2) consisted of a dictionary-matching approach using the dictionary provided by LINNAEUS (18), with post-processing rules to remove ambiguities. For the normalization of gene/protein names (Task 3), the system applied a dictionary lookup strategy where two gene dictionaries were checked in sequence. The dictionaries were created from the BioLexicon gene dictionary (19), the first one containing only the preferred name of each gene, and the second one containing all the synonyms. Species annotations were used to filter out ambiguous genes, so that an ambiguous gene mention is assigned the gene identifier according to the nearest species recognized in the same sentence, passage and/or full-document context. When no organism mention was found, the human gene identifiers were kept.
Argo
Team members: Riza Batista-Navarro, Jacob Carter and Sophia Ananiadou.
Argo is a workbench for building text-mining solutions with a rich graphical user interface (14). Central to Argo are customizable workflows that users compose by arranging available elementary analytics to form task-specific processing units. For the BioC task, Batista-Navarro et al. focused on developing new methods for the recognition and normalization of mentions denoting genes/proteins and organisms. These methods were trained on these corpora: the CHEMDNER corpus of patents containing gene/protein name annotations (20), and the S800 corpus of PubMed® abstracts annotated for organism mentions (21). The Argo system leveraged these previously developed tools for data pre-processing: the LingPipe sentence splitter for detecting sentence boundaries (http://alias-i.com/lingpipe), OSCAR4’s tokenizer for segmenting sentences into tokens (22) and the GENIA Tagger for lemmatization as well as part-of-speech and chunk tagging (23). The recognition of gene/protein (F-score 70%) and organism mentions (F-score 73%) in text was addressed by training Conditional Random Fields models on lexical and syntactic features extracted by the above pre-processing tools over the training corpora, combined with semantic features drawn from dictionary matches. To facilitate the normalization of the recognized mentions, the team developed rules based on string similarity, exploiting the Jaro-Winkler and Levenshtein distance measures.
NTTMUNSW
Team members: Onkar Singh, Jitendra Jonnagaddala, Hong-Jie Dai and Emily Chia-Yu Su.
Singh et al. (15) developed three BioC-compatible components for processing the full text articles in BioC format, and generated annotation results for species and gene/protein names along with their NCBI Taxonomy IDs and Entrez Gene IDs. The preprocessing NLP pipeline included: sentence splitting, tokenization, part-of-speech tagging and abbreviation recognition. The gene mention recognizer, a conditional random fields model, was trained on the generated linguistic information and these features were used as the input for the species mention recognizer. In addition to the full species terms, the species recognition component can recognize prefixes in a gene name that refer to a species (F-score 94% on IGN corpus). For instance, the prefixes ‘h’, ‘Hs’, ‘Sc’ and ‘Ca’ in the gene mentions ‘hLysoPLA’, ‘HsUap1p’, ‘ScUAP1’, ‘CaUap1p’ are recognized by this module, and represent ‘Homo sapiens’, ‘Saccharomyces cerevisiae’ and ‘Candida albicans’, respectively. The team also developed a multistage system optimized for processing full-text articles (using the BioCreative II.5 corpus). The multistage algorithm uses the research article structure, and follows the general distribution of gene/protein-related information throughout the article to assign information accurately to each section. For example, the introduction section generally contains the key genes described in the article, and these genes when repeated in the Results section are usually mentioned in their abbreviated form. Their module parses the articles in a predefined way, and remembering the already seen instances, has an increased accuracy.
Molecular interaction recognition systems
The following teams contributed runs for molecular interaction passage recognition (Tasks 4, 6 and 7). These teams made use of the output produced by the previous teams, as the knowledge of genes/proteins and species in the text is necessary and informative for the predictions in these tasks. The outputs of all teams were submitted to the task organizers and they were processed to compute one integrated output for presentation to the curators. As mentioned earlier, we merged Task 5 with Task 7 after receiving task proposals from participating teams.
PIPE
Team members: Yung-Chun Chang, Yu-Chen Su, Chun-Han Chu, Chien Chin Chen and Wen-Lian Hsu.
Chang et al. (24) contributed one prediction output for Task 4, detection of full-text passages mentioning protein–protein interactions. In this work, three PPI corpora: LLL (25), IEPA (26) and HPRD50 (27) are used to train and develop the method. The PPI extraction system consists of three main components: interaction pattern generation, interaction pattern tree construction and convolution tree kernel. Prior knowledge of PPI was first used to mark the words in a given corpus, and frequently co-occurring tuples were collected to generate interaction patterns via a Probability Graphical Model. Afterwards, a PPI sentence was represented with the interaction pattern tree structure, which is the shortest path-enclosed tree of the instance enhanced by a rewriting procedure. Finally, a convolution tree kernel was employed to capture the structured information in terms of substructures and determine the similarity between sentences. Results showed this method was effective in extracting PPI and achieved about 70% F1-Score.
Rule-basedextendeddependency graph method for prediction of PPI mentions
Team members: Yifan Peng, Cecilia Arighi, Cathy H. Wu and K. Vijay-Shanker.
Peng et al. (28) also worked on Task 4, and contributed four different outputs for different settings of their method. Their system of detecting passages describing protein–protein interactions in full text articles utilized the Extended Dependency Graph as an intermediate level of representation to abstract away syntactic variations in the sentence (29). This method made full use of all gene/protein and species annotations of previous teams, and used open source available tools such as the Bllip parser (30) to obtain parse trees, and CCProcessed from Stanford tools (31) to extract dependencies. As a result of the Extended Dependency Graph construction, the team created three basic predicate-argument rules to extract PPI pairs in sentences. The team labeled a sentence as positive if it contained PPI pair(s), and two additional rules were used to detect additional passages with PPI pairs. Experiments on 20 in-house annotated full-text articles showed an F-value of 80.5. Using only the three basic rules, experiments on AIMed (32) further confirm that the proposed system can achieve an F-value of 76.1 for sentence selection and an F-value of 64.7 for unique PPI detection.
Extraction ofpassages with experimental methods for physical interactions
Team members: Ferhat Aydın, Zehra Melce Hüsünbeyi and Arzucan Ozgür.
Aydın et al. (33) developed a system for identifying evidence passages for protein–protein interactions in full-text articles based on information retrieval techniques. In addition, their method was refined for the most frequent experimental methods, and they provided an extra tag that identified the precise method of interaction. The team started by selecting the most frequent experimental methods in the BioCreative III IMT (Interaction Method Task) data set and manually annotated a set of 13 full text articles containing these methods in BioC format. This data set was used for system development. Their approach is based on generating queries for each experimental method by making use of the Proteomics Standards Initiative-Molecular Interactions (PSI-MI) ontology and the manually annotated articles. The terms and synonyms of the method in the PSI-MI ontology were used for query generation. The tf–rf (term frequency–relevance frequency) weighting metric was used to rank the words in the training documents and determine the salient keywords for each experimental method. The salient keywords were used for query expansion. Given a test document, the queries for all experimental methods were run, and sentences that produced a similarity score higher than a threshold value for an experimental method were selected as relevant (F-score 63%). The team submitted two runs on the evaluation set of 120 articles.
Identification of protein–protein interaction passages and methods passages
Team members: Sun Kim and W. John Wilbur.
Kim and Wilbur submitted three runs for PPI passages and PPI method passages. The contribution of PPI passages is based on the output from PIE the search (34). The PIE engine utilizes a support vector machine (SVM) classifier with multiword, substring, MeSH® term and dependency relation features. Although the system was designed for a PPI document triage task, the team found that it worked reasonably well at identifying PPI informative sentences. The following two methods were developed with the goal of identifying passages containing experimental methods for PPI. The first method incorporated distant supervision (35) into a classification task. First, candidate sentences with GI pairs (appearing in BioGRID) were obtained from 1347 PubMed Central® (PMC) articles. There was no overlap between these articles and the dataset used for the BioC task. Among the candidates, sentences with a greater than 5.0 tf·idf score were selected based on BioGRID genetic interaction descriptions, and were labeled as negatives for PPI interaction. This set was merged with the PPI interaction method dataset from BioCreative II (8). The combined set contained 21828 sentences, and was used to train an SVM classifier for PPI method prediction. The team found that the distant supervision approach was highly effective for accepting PPI, but also rejecting GI sentences. The second method began by collecting names from the 17 methods contained in the BioGRID descriptions of methods for PPI detection. Each name was used to search for literature containing the name to find recent review articles describing the method, facets of the method, or updates to the method. The search was performed in two ways. First, all the PubMed documents containing the name were used to perform machine learning to recognize literature that would be likely to contain the name and then the highest scoring documents in PubMed based on this learning were examined. Second, the Google search engine was used to access literature to look for useful top scoring documents. By examining these documents 415 one and two token phrases describing PPI experimental methods were manually collected. Any sentence was then scored by how many of these phrases were found in the sentence. This simple sentence ranking scheme was applied to articles believed to contain sentences describing experimental methods for PPI detection. In this selected set of documents, these methods proved useful.
Identification of genetic interaction evidence passages
Team members: Rezarta Islamaj Doğan, Sun Kim, Andrew Chatr-Aryamontri, Donald C. Comeau and W. John Wilbur.
Islamaj Doğan et al. (36) contributed four different runs for Task 7. This particular task was novel as there existed no prior work, and no prior data to help build a new system. In addition, text describing genetic interactions is difficult to identify due to lack of a simple definition for these interactions. This team therefore prepared two manually annotated datasets: 1793 sentences from PubMed abstracts and 1000 sentences from full text articles. The sentences in the first dataset were annotated for gene, organism and chemical entities, as well as for trigger words describing gene and gene function modifications and observed phenotypic effects of a genetic interaction. The sentences in the second dataset were marked yes or no, depending on whether they described a genetic interaction or not. Furthermore, the team built two classification systems to identify genetic interaction evidence (F-score 74%), a context-feature based SVM using word and context features, and an information-retrieval based SVM using Lucene (https://lucene.apache.org) to obtain feature values. SVM features were phrases describing gene function and interaction evidence (e.g. double mutant analysis and synthetic lethality). The value for a feature as applied to a given sentence is determined using the feature (phrase) as a query in Lucene to score against that sentence. Both models were applied to the BioC track dataset and results were submitted for inclusion in the complete BioC Track system.
System prediction
Each BioC task had multiple submissions from participating teams, but only one prediction set could be shown to BioGRID curators for evaluation. Thus, we needed a process to merge submitted runs. To achieve this goal, we first created a manually annotated set, and applied a machine learning approach to combine submitted outputs.
Dataset
For merging multiple outputs from teams and evaluating the biocurator assistant system, we recruited four curators from BioGRID to build a gold-annotation set. Since most of the information in the BioGRID database was from the yeast or human domain, we randomly chose 60 full-text PMC articles for each of these organisms. For the selected documents in the human set, there were 38 Open Access PMC articles (OAPMCs) with entries in BioGRID containing both PPI and GI information, 17 OAPMCs with PPIs and 5 OAPMCs with GIs. Table 4 summarizes the newly created annotation set for this merging and evaluation process.
Evaluation set used for optimizing the merger of submitted runs
| Organisms | Documents | Molecular interaction information |
|---|---|---|
| Yeast | 60 | PPI and GI |
| Human | 38 | PPI and GI |
| Human | 17 | PPI |
| Human | 5 | GI |
Documents were randomly selected from PMC articles relevant to either yeasts or humans. Of these, 98 documents contained both PPI and GI information, the remaining 22 documents contained either PPI or GI.
As shown in Figure 3, BioGRID curators used an interface to annotate gene/protein/organism mentions and PPI/GI passages. This annotation process was straightforward: first, highlight a piece of text and second, select one of the annotation types. For gene/protein/organism mentions, curators were asked to enter a gene/taxonomy ID before selecting an annotation type. It is time-consuming to curate all gene/protein names and their corresponding IDs in a full-text article, hence curators were asked to annotate only gene/protein/organism names appearing in PPI/GI passages. Due to the tight schedule, the 120 full-text articles were divided into four sets and each set was assigned to one curator. After the BioCreative Workshop, the task organizers and the BioGRID curators have worked closely together to correct and refine the original annotations and have produced a true gold standard corpus of molecular interaction names and passages useful for curating the 120 full text articles. This work is described elsewhere (37).

Annotation Interface for full-text PMC articles. This is a screenshot of our annotation interface that curators used to create a gold-standard annotation set. For annotations, a curator selects relevant text and chooses an annotation type button on the screen. Gene ID and Tax ID options are for assigning IDs to gene and organism names.
Merging process for submitted predictions
To evaluate the biocurator assistant system, we first selected 10 articles for each curator from the annotated full-text set, and assigned them as a test set (i.e. the test set contained 40 articles in total). The test set articles for each curator were chosen from among the full-text articles that curator had not seen during the initial annotation process. Then, for each curator, the remaining 110 articles were used as a training set to optimize parameters for merging the outputs of the submitted runs on their 10 article test subset. Here we describe this process, which was somewhat different for entity recognition tasks and the interaction recognition tasks.
For gene/protein/organism tasks (Tasks 1, 2 and 3), we measured the performance of team submissions by precision and overlaps between submitted runs. Since all teams performed reasonably on these tasks, the merging process for Tasks 1, 2 and 3 did not include machine learning and simply took the union of submitted runs to maximize recall. This is a reasonable strategy for NER tasks because curators prefer high recall.
The PPI/GI tasks are somewhat different than NER tasks. Users expect high recall in general, meanwhile precision should not be ignored. In order to merge the results, we tried voting and SVM learning using binary scores as features. The results were not satisfactory. Thus, we created an SVM-based approach for each submission that used the annotations of that submission to assign a score for each sentence (Figure 4). A final SVM used these scores as features for merging and optimizing the results. Here are the detailed steps.
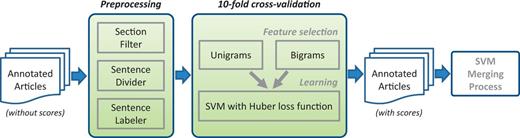
Score assigning process for each submission from PPI/GI tasks.
Score assigning process
Considered all data in the 120 articles.
Treated each sentence of the text individually.
Removed sentences in uninformative sections such as acknowledgements and references.
For each submitted run for each team, assigned a score to each sentence based on that individual submission’s predictions.
Treated a submission’s predicted sentences as a gold standard.
Used unigrams and bigrams from text as features and trained an SVM classifier (38) to identify the predicted sentences of this submission from the rest.
Performed a 10-fold cross-validation to learn the best weights.
Submission merging process
After this merging and optimization process, the 40 system annotated articles were uploaded to the BioC viewer (http://viewer.bioqrator.org) (39) for evaluation, and each curator curated PPI and GI pairs from his/her 10 assigned articles.
For each curator, for all sentences in the corresponding training set, we learned an SVM classifier using the obtained scores above as feature weights. If there are four runs for a task, the number of features is exactly four, i.e. for each training sentence, there are four feature values from four submissions. This process prioritizes submitted runs while maximizing the prediction performance.
Made predictions on each 10 article test set using the combined results produced by the final SVM.
Results and discussion
After testing the system, BioGRID curators were asked to rate the usefulness of the system and its functionality on a scale of 1 (bad) to 5 (good) and were encouraged to provide feedback about aspects that would benefit from further improvement. Table 5 presents the questionnaire used for feedback. The questionnaire consists of seven categories: ‘Overall reaction’, ‘Overall comparison to similar text mining-based curation systems’, ‘System's ability to help complete tasks’, ‘Prediction performance’, ‘Design of BioC Viewer’, ‘Learning to use BioC Viewer’ and ‘Usability’. In the table, average ratings from the four curators are shown for each question. The rating with ‘N/A’ (not available) was not used for calculating average rates. From the table, the curators were positive overall for the design (V) and the learnability (VI) of the curation system. Only two curators had experience on other text mining tools and their responses were positive as well (II). However, passage predictions still need improvement in accuracy to significantly support the curation process. Other comments noted that functionalities for the actual curation were limited. This shows that curators are interested in having the text mining functionalities incorporated in their systems for easier and better curation. The BioC biocuration assistant tool interface was designed as a viewer, and this can be changed by incorporating the curators' comments and suggestions in the future. All responses and comments from BioGRID curators are available in Supplementary material.
Questionnaire used for user feedback
| Questions | Rates |
|---|---|
| I. Overall reaction | |
| a. Please rate your experience with BioC Viewer. | 3.3 |
| b. Overall, I am satisfied with BioC Viewer. | 3.0 |
| c. I would recommend BioC Viewer to other PPI/GI curators. | 2.8 |
| II. Overall comparison to similar text mining-based curation systems | |
| a. It is easy to use BioC Viewer. | 5.0 |
| b. I am satisfied with using BioC Viewer. | 4.0 |
| c. BioC Viewer is powerful enough to complete the task. | 3.0 |
| III. System’s ability to help complete tasks | |
| a. Speed: the system would reduce annotation time to reach my curation goal. | 3.5 |
| b. Effectiveness: the system would help me get closer to my curation goal. | 3.0 |
| c. Efficiency: I can be both fast and effective with the system. | 2.8 |
| IV. Prediction performance | |
| a. Task 1 (gene/protein NER) | 4.3 |
| b. Task 2 (organism NER) | 2.7 |
| c. Task 3 (gene/protein name normalization) | 3.8 |
| d. Task 4 (Passages with PPIs) | 3.3 |
| e. Task 6 (Passages with PPI experimental systems) | 2.5 |
| f. Task 7 (Passages with GI types) | 3.0 |
| V. Design of BioC Viewer | |
| a. It was easy to find and read information. | 4.0 |
| b. Highlights were adequate and helpful. | 3.5 |
| c. Information was well organized. | 3.5 |
| VI. Learning to use BioC Viewer | |
| a. It was easy to learn how to operate the interface. | 4.3 |
| b. It was easy to remember features in BioC Viewer. | 4.3 |
| c. It was straightforward to use the interface. | 4.3 |
| VII. Usability | |
| a. The interface was fast enough to do my job. | 3.5 |
| b. The interface was performed consistently. | 4.0 |
| c. The interface provided a means to easily correct mistakes. | 3.0 |
For each question, BioGRID curators rated on a 1 (bad) to 5 (good) scale. The scores shown are the average rates from four curators.
Figure 5 depicts the detailed ratings from curators for ‘Prediction performance’. Curators were satisfied with gene/protein NER and normalization (Tasks 1 and 3) overall, whereas they showed less favorable views for the organism NER and normalization task (Task 2). Curator 4 also did not assign any score for Task 2. This may be partly because the goal was to curate PPI and GI pairs, not organism mentions. Organisms are only considered as a part of the normalization process for gene/protein names and are not included in BioGRID.
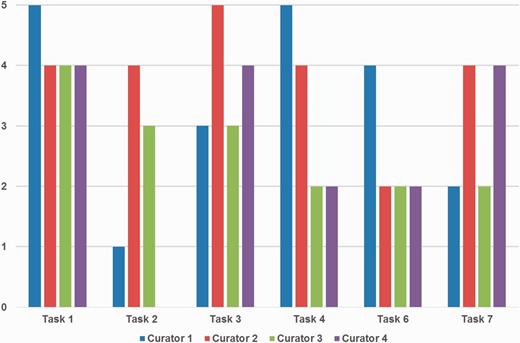
Curators’ ratings for prediction performance for each task. Tasks 1, 2 and 3 are gene/protein named entity recognition (NER), species/organism NER and gene/protein name normalization, respectively. Tasks 4, 6 and 7 are passages with protein–protein interactions (PPIs), PPI experimental methods and genetic interaction types, respectively. Tasks 1 and 3 received positive responses overall, however ratings were mixed for other tasks depending on curators’ preferences. Curator 4 did not assign a score for Task 2.
The PPI/GI passage tasks received rather mixed ratings, and the reactions for finding passages with PPIs and GI types (Tasks 4 and 7) were slightly better than finding passages with PPI experimental methods (Task 6). Curators' comments suggest that this could be a matter of personal display preference, i.e. some may prefer higher recall, but others may prefer higher precision. This suggests that an adjustable function to adjust the prediction output on display is desirable and may be a useful feature to add to the biocurator assistant system.
Conclusions
The purpose of the BioC track in BioCreative V was to create a set of complementary modules that could be seamlessly integrated into a system capable of assisting BioGRID curators. Specifically, the resulting interactive system triaged sentences from full text articles in order to identify text passages reporting mentions and experimental methods for protein–protein and genetic interactions. These sentences were then highlighted in the biocuration assistant system for curators. This task was unique since participating teams had to produce independent, but collaborative modules for the biocuration assistant system. The task was divided into several smaller tasks that required the identification of passages or sentences describing genes/proteins/species involved in the interaction as well as mentions and experimental methods for describing molecular interactions. Nine teams, world-wide, developed one or more modules independently, to address the challenges as outlined by the specific subtasks.
The most important achievement of this task is undoubtedly the achievement of interoperability. Data was received, produced and exchanged in BioC, which was easy to learn and simple to use. This simple format avoided many interoperability hurdles. The organizing team also developed a machine learning process to merge 24 submissions from collaborating teams. Through an evaluation process, four BioGRID curators judged the integrated output and the biocuration assistant system in terms of its practical usability. Their feedback indicated that the performance of the text mining results for gene/protein NER and normalization was adequate to support the biocuration task. Further work remains to be done for improving suggestions of molecular interaction evidence passages. The curators gave positive feedback regarding the user-friendliness and the biocuration assistant system in general. Future work may also include improving the curation tool based on curators' feedback. The dataset annotated during the execution of this task will also be made available to the community to encourage development and improvement of text mining systems assisting biocuration.
Acknowledgements
The authors would like to thank Cecilia Arighi for providing resources for the user questionnaire.
Funding
Intramural Research Program of the NIH, National Library of Medicine to S.K., R.I.D., W.J.W. and D.C.C.; National Institutes of Health [R01OD010929 and R24OD011194] to K.D and M.T.; Genome Quebec International Recruitment Award to M.T.; FCT—Foundation for Science and Technology under the FCT Investigator program to S.M.; The European Union’s H2020 project MedBioinformatics (Grant Agreement No 634143) to D.C.; National Science Foundation [DBI-1062520] and the National Institute of Health [DE-INBRE P20GM103446] to Y.P., C.A., C.H.W. and K.V.; Marie Curie FP7-Reintegration-Grants within the 7th European Community Framework Programme to F.A., Z.M.H. and A.Ö.; Basic Science Research Program through the National Research Foundation of Korea [2012R1A1A2044389 and 2011-0022437] to D.K. Funding for open access charge: Intramural Research Program of the NIH, National Library of Medicine.
References
Author notes
Citation details: Sun Kim, S., Doğan, R.I., Chatr-Aryamontri, A. et al. BioCreative V BioC track overview: collaborative biocurator assistant task for BioGRID. Database (2016) Vol. 2016: article ID baw121; doi:10.1093/database/baw121
These authors contributed equally to this work.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}