





Abstract
The lack of interoperability among biomedical text-mining tools is a major bottleneck in creating more complex applications. Despite the availability of numerous methods and techniques for various text-mining tasks, combining different tools requires substantial efforts and time owing to heterogeneity and variety in data formats. In response, BioC is a recent proposal that offers a minimalistic approach to tool interoperability by stipulating minimal changes to existing tools and applications. BioC is a family of XML formats that define how to present text documents and annotations, and also provides easy-to-use functions to read/write documents in the BioC format. In this study, we introduce our text-mining toolkit, which is designed to perform several challenging and significant tasks in the biomedical domain, and repackage the toolkit into BioC to enhance its interoperability.
Our toolkit consists of six state-of-the-art tools for named-entity recognition, normalization and annotation (PubTator) of genes (GenNorm), diseases (DNorm), mutations (tmVar), species (SR4GN) and chemicals (tmChem). Although developed within the same group, each tool is designed to process input articles and output annotations in a different format. We modify these tools and enable them to read/write data in the proposed BioC format. We find that, using the BioC family of formats and functions, only minimal changes were required to build the newer versions of the tools. The resulting BioC wrapped toolkit, which we have named tmBioC, consists of our tools in BioC, an annotated full-text corpus in BioC, and a format detection and conversion tool.
Furthermore, through participation in the 2013 BioCreative IV Interoperability Track, we empirically demonstrate that the tools in tmBioC can be more efficiently integrated with each other as well as with external tools: Our experimental results show that using BioC reduces >60% in lines of code for text-mining tool integration. The tmBioC toolkit is publicly available at http://www.ncbi.nlm.nih.gov/CBBresearch/Lu/Demo/tmTools/.
Database URL:http://www.ncbi.nlm.nih.gov/CBBresearch/Lu/Demo/tmTools/
Introduction
There is an increasing demand of text-mining tools in the biomedical and life sciences domain. Many recent BioNLP challenge tasks (1–5) are focused on extracting structured information from scientific articles and clinical notes. Research groups around the world are developing a variety of stand-alone text-mining tools. Typically, a tool is developed using certain data representation and programming conventions as preferred by the individual research group. To build complex text-mining applications or pipelines, it is often required to combine multiple tools, possibly designed by different groups. The current practice of independent tool development poses a hindrance to tool interoperability and integration. To use a new tool or a new data set, text-mining researchers spend a substantial amount of time developing algorithms for processing the new data format. This heterogeneity in data representation slows down the development of powerful applications and thereby leads to inefficiencies in research and innovation.
There have been quite a few efforts to promote interoperability among text analytics tools and data sets. The BioCreative MetaServer (6) is designed to combine natural language annotations produced by different groups; any group interested in contributing to the central server is required to implement an annotation server based on a predefined three-layered framework consisting of data, communication and application layers. Unstructured Information Management Architecture (UIMA) (7, 8) and General Architecture for Text Engineering (GATE) (9) are two notable proposals that prescribe using a predefined framework to develop text-mining applications to achieve interoperability among independently developed tools. Although type systems such as U-Compare (10) and technical utilities such as UIMAFit (11) have been developed to facilitate easier integration of UIMA-compliant tools, the development of a UIMA- or GATE-compliant application requires the entire tool to be (re-)written into framework-specific constructs and presents a steep learning curve (12). Another approach to accomplish interoperability among annotated corpora is to promote a common output data format. For example, a recent effort in this direction, BioC (13), is based on a minimalist approach in that it offers interoperability by stipulating minimal changes in existing applications or data sets. The goals of BioC are simplicity, reusability, interoperability and wide use. In a nutshell, BioC is a family of XML formats that define how to present text documents and annotations. BioC is different from previously proposed formats, such as IeXML (14, 15), in that BioC also provides tools to read and write documents in the BioC format in multiple programming languages to further minimize the efforts of tool developers.
In this article, we present our efforts on using BioC to repackage the suite of text-mining software and Web-based tools (16–22) developed by the biomedical text-mining group at the National Center for Biotechnology Information (NCBI). Specifically, we wrap five stand-alone biomedical named entity recognition (NER) tools, one Web-based annotation tool and one annotated text corpus, into BioC. In addition, we provide a format converter tool that allows conversions among three different data formats: two tool-specific formats and BioC. By making our toolkit BioC-compatible, we expect to enhance its interoperability and the capacity to develop more sophisticated text-mining applications such as extracting relations between entities (23, 24). We evaluate the BioC-compatible toolkit at two levels: (i) the ability to integrate our tools with each other and (ii) the ability to integrate our tools with tools developed by other groups. This study was conceived and conducted through our participation in the BioCreative IV interoperability task (25, 26).
Method
In this section, we first introduce our toolkit that comprises six tools for concept recognition and annotation (shown in Figure 1), and an annotated text corpus for Gene Ontology (GO) concept recognition. Then, we describe the key steps and challenges in creating a BioC compatible version of the tools and the text corpus.
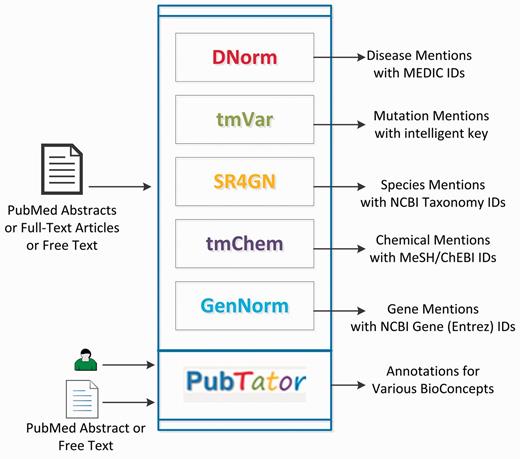
Visual summary of our text-mining toolkit.
PubTator
We have developed a Web-based annotation tool called PubTator (20, 27, 28) for assisting manual curation. PubTator was developed using JavaScript, Perl-CGI, HTML and MSSQL. PubTator is synchronized with PubMed® and supports semantic annotation and search of key biomedical entities and their relationships in PubMed articles. Similar to Entrez Programming Utilities (29), PubTator also offers programmatic access to its results with a list of parameters specifying the output content and format: (i) a list of PMIDs to be retrieved, (ii) Bioconcepts (‘Gene’, ‘Chemical’, ‘Disease’, ‘Mutation’ and ‘Species’), to be included and (iii) the output format (See Table 1).
Input/output formats supported by our text-mining toolkit
| tmTools | Formats supported (I = Input, O = Output) | ||||
|---|---|---|---|---|---|
| PMC XML | Free text | Tool-specific format 1a (PubTator) | Tool-specific format 2b (GenNorm) | Tool-nonspecific format (BioC) | |
| tmChem | I | I | I/O | ||
| DNorm | I | I | I/O | ||
| tmVar | I | I | I/O | I/O | |
| SR4GN | I | I | O | I/O | |
| GenNorm | I | I | O | I/O | |
| PubTator | I | I | I/O | I/O | |
Concept recognition tools
We have developed several NER tools for automatically recognizing key biomedical concepts, such as chemicals, diseases, genes, mutations and species, from scientific publications (30, 31). Each tool accepts a PubMed or PMC full-text article or free-text as an input and identifies the biomedical entities at both mention and concept levels.
DNorm (1, 19) is an open-source software tool to identify and normalize disease mentions from biomedical texts. DNorm is based on the pair-wise learning to rank algorithm and is the first technique to use machine learning for disease normalization. This tool was developed in Java.
tmVar (18) is a machine learning system for mutation recognition to assist biomedical curation. It is based on conditional random fields and identifies many types of mutations and sequence variants in protein, gene, DNA and RNA levels for biomedical curation. This tool was developed in Perl and uses the CRF++ module developed in C++.
SR4GN (16) is a species recognition tool optimized for the gene normalization task. It is a rule-based system that identifies species from full-texts and pairs them with corresponding gene or protein mentions. This tool was developed in Perl.
tmChem (21) is a machine learning-based NER system for chemicals. The system is designed to identify and normalize a wide variety of chemical mentions in literature, including identifiers, brand and trade names and also systematic formats. The system uses conditional random fields with a rich feature set and rule-based post-processing modules for resolving local abbreviations and improving consistency. This tool was developed in Java.
GenNorm (17) is a rule-based tool for gene name recognition, species assignation and species-specific gene normalization. GenNorm addresses the challenging issues of orthologous gene ambiguity and intra-species gene ambiguity. This tool was developed in Perl.
The BC4GO corpus
More recently, we developed the BC4GO corpus (22) (not shown in the figure), a corpus of 200 full-text articles along with their GO annotations describing genes and gene product attributes across species and databases. As annotations, the corpus presents the evidence sentences along with the gene/protein entities, GO terms and GO evidence codes. The corpus was developed with eight expert biocurators using a Web-based annotation tool. This is the official corpus for the BioCreative IV Track-4 GO Task (32), which tackles the challenge of automatic GO annotation through literature analysis.
Building BioC compatible tools
The BioC family of XML formats and functions comprises the following four items:
The XML Document Type Definition (DTD) that defines the syntax, i.e. how to present text document and annotations (with global offsets) to share common information. The BioC format allows standoff annotations in two ways where users can choose to keep one file or two separate files (for articles and annotations, respectively).
A key file to describe the semantics, i.e. how to interpret the data (XML elements) in the BioC annotation file. BioC allows many different kinds of annotations to be represented, including title, abstract, sentences, paragraphs, biomedical entities (e.g. gene), etc. A key file is custom-created for a specific type of problem domain.
C++, Java, Ruby, Python, Go and SWIG libraries that include functions and classes to read and write documents in the BioC format and to hold the documents in memory.
A format converter to translate an article from the PMC (or PubMed) XML format to the BioC format.
To comply our tools with BioC, we modified the input and output formats of the tools by adding BioC as a new option, and translated the articles and the annotations in our corpus to corresponding BioC files. Table 1 summarizes the formats originally supported by the tools.
PubTator
The original input/output format for PubTator was a predefined format, the tool-specific format 1. To make PubTator BioC compatible, we added a new format option giving users the option to input and output in the BioC format. For our purposes, we chose to keep the articles and annotations into a single file so that users need to upload a single file when importing annotations into PubTator. We defined the tmBioC.key file (submitted as Supplemental Data) that describes specific attributes such as bioconcept types, database identifiers, locations and mentions.
Concept recognition tools
The primary effort in converting the various concept recognition tools to BioC was to define an appropriate key file. As the semantics of all these tools were similar to PubTator in terms of the type of data, we used the same key file, tmBioC.key, which was used for BioC compatible version of PubTator. The same key file was used for interpreting the input articles/abstracts and the output articles/abstracts with annotations. Once the key file was finalized, the remaining efforts were to add a new input/output format (i.e. BioC) to the tools.
The mutation recognition tool, tmVar, originally accepted the tool-specific format 1, free text and the PMC XML format. The output format was the tool-specific format 1. For GenNorm and SR4GN, the input formats were free text, PMC XML format and the tool-specific format 2, and the output format was the tool-specific format 2. Translation of these tools into respective BioC versions required additional efforts, as these tools were authored in Perl and the BioC functions were only available in Java and C++ when we conducted this study. Hence, a Perl implementation of BioC functions was implemented for the purpose of this study. Finally, to develop the BioC versions for tmVar, GenNorm and SR4GN, we added the BioC format as a new option for input/output. The key difference between the tool-specific format 2 and the BioC format is that while BioC recommends specifying global offsets, the tool-specific format 2 calculates offsets for separate sentences. Also, the tool-specific format stipulates separate files not only for articles and annotations but also for different kinds of bioconcepts. Hence, the offset calculation and file writing functions for GenNorm and SR4GN tools had to be modified accordingly.
The previous output format for tmChem was the BioCreative IV CHEMDNER format, which is essentially the same as the tool-specific format 1, representing one NER mention per line. DNorm is a relatively new tool and did not previously have a default output format. As both tools are built on top of BANNER (33), input compatibility with BioC only required adding the new data set loading class, provided by the BioC family of tools, in BANNER. Modifying the output required modifying the class containing the main method to output the BioC format.
BC4GO corpus
First, the 200 full-text articles of the BC4GO corpus were uploaded to the annotation tool, and eight annotators performed annotation. In the meanwhile, the PMC XML data model formats of those articles were converted to the BioC format using the format converter provided by the BioC tools.
Then, we downloaded the HTML files, and extracted the annotations including annotation texts, genes, GO terms and identified their offsets. There were certain challenges in creating the annotation files that required additional programming efforts. The first challenge was in creating the BioC annotation file using the user annotations downloaded from the Web-based annotation tool. We observed encoding discrepancies in the PMC XML file and the downloaded file. The original article was published in Unicode; the file in PMC XML format was encoded in UTF-8 but was converted to ASCII in the BioC format. Although the annotation results downloaded from the Web were also encoded in ASCII, they were translated using a different Unicode-ASCII converter table. For example, the term ‘neurexin-1α’ (see PMID: 22262843 in corpus) would read ‘neurexin-1alpha’ in the PMC XML file but ‘neurexin-1I+-’in the downloaded annotation file. To maintain consistency between the BioC article and annotation files, we converted the characters using a neighbor matching method as described in (22).
Another challenge was presenting the evidence sentences that contain multiple discontinuous sentences, possibly from different passages in the article. For example, in PMID 19321442, the curator annotated ‘Surprisingly, pep2-SVKI, which blocks both PICK1 and ABP/GRIP PDZ domains, did not completely block the decrease in the rectification index (Figure 2C, pre-OGD = 0.42 ± 0.04, post-OGD = 0.37 ± 0.03, n = 6, P < 0.05).’ from the fourth paragraph in the RESULTS section, and ‘We observed a time-dependent reduction in GluR2 surface expression by OGD (Figure 3B), which is consistent with a previous study’ (17) from the sixth paragraph in the same section, as the evidence sentence for GO: 0009986 (cell surface). The two sentences appeared in different passages in the BioC format article, i.e. belonged to different sub-nodes in the XML file. We addressed this challenge by linking these evidence sentences using the same annotation ID, i.e. they are treated as one whole evidence sentence for a GO term.
Finally, for each article we created a corresponding BioC annotation file for the associated GO annotations using the tools provided by BioC. For the gene entity, we provide both the gene mention as appeared in text and its corresponding NCBI Gene identifier. In the BioC released corpus, each article is named by its PubMed identifier, e.g. ‘20130316.xml’. The annotation file associated with the article file shares the same PMID in the file name, e.g. ‘annotation_20130316.xml’. The annotation file includes all annotations of the article; each annotation has a unique ID and is defined by four distinct elements: gene, go-term, go-evidence and type. We define separate key files to describe the full-text articles and the annotation files with GO annotations, namely pmc_go.key and go_annotation.key, respectively.
One limitation of the corpus released in BioC is that the BioC annotation file of an article would not contain an evidence sentence that is located in the ‘Acknowledgement’ section of the article (see PMID 18695045) because this section is not provided in the original PMC XML file for the article. Also, in some cases, such as line feeds and hyperlinks, incomplete sentences were created because of the additional space characters in the original PMC XML files. Such cases were manually processed to create consistent BioC annotation files.
Format converters
The BioC family does provide a format converter to translate articles in the PMC XML format to the BioC format. However, there is no converter available to translate the annotation files. Hence, we developed a Perl-based format converter that allows automatic detection and format conversion among three different data formats: tool-specific formats 1 and 2, and BioC. A major issue in implementation of the converter was the discrepancies in file saving conventions adopted by different formats. For a given article with annotations, the tool-specific format 2 generates separate files for the article and for different kinds of bioconcepts such as genes, species, chemicals, etc. In contrast, both tool-specific format 1 and BioC formats prescribe saving sentences and annotated mentions into a single file. Considering these discrepancies, we developed and wrapped multiple small format detectors and converters into a single tool that allows users to translate annotations for a given article into a desired output format.
Results
Our upgraded toolkit, which we have named tmBioC, comprises the new BioC versions of all tools, the common tmBioC.key file, and the format converter. tmBioC is made publicly available at http://www.ncbi.nlm.nih.gov/CBBresearch/Lu/Demo/tmTools. The key file is provided as a Supplementary Data.
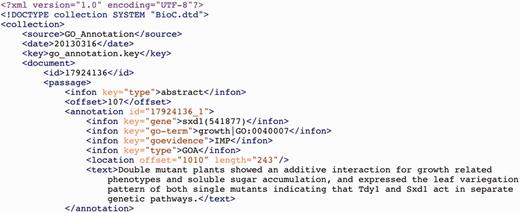
To describe the outputs of our concept recognition tools, we use a PubMed abstract (PMID 20085714) that contains mentions of multiple biomedical entities, including genes, mutations, chemicals and diseases, as a running example. A snippet of the BioC input file for this example is shown in Figure 2, and a snippet of BioC output file showing the integrated results from all the tools are displayed in Figure 3. The BioC annotation file contains detailed information about each annotation. For example, the annotation id ‘11’ is represented by the mention ‘c.95de1T’ (text tag) and located in the global offset 679 with a length of 8 characters (location tag). Also, this annotation is a mutation type annotation (infon key = ‘type’) and is normalized to an automatically generated intelligent key, ‘c|DEL|95|T’ (infon key = ‘tmVar’) that captures the fine grained mutation information in the format<Sequence type, mutation type, wild type, mutation position, mutant>. As another example, the annotation id ‘17’ is presented by the mention ‘cyclic AMP’ and located in the global offset 919 with a length of 10 characters; this annotation is a chemical type and is normalized to a concept in the MeSH vocabulary, ‘D000242’.

A snippet of the BioC article file for PMID 20085714.

A snippet from the BioC annotation file for PMID 20085714 (integrated result of applying our five concept recognition tools on the abstract).
The BioC version of the BC4GO corpus, with 200 BioC article files and 200 BioC annotation files, can be downloaded at the BioCreative IV Track 4 task’s official webpage, http://www.biocreative.org/tasks/biocreative-iv/track-4-GO/. The key files, pmc_go.key and go_annotation.key, were submitted as part of the BioCreative IV Track 1 submission. These key files are also provided as Supplemental Data. A snippet of the BioC annotation file corresponding to the PubMed article with PMID 23840682 is shown in Figure 4.
A Snippet from the file annotation_23840682.xml from the BC4GO corpus
Interoperability evaluation
To demonstrate the interoperability of tmBioC, we conducted two experiments, with internal and external tools, respectively. For studying the interoperability of tmBioC within the toolkit, we integrated PubTator with all the five concept recognition tools, tmVar, tmChem, GenNorm, SR4GN and DNorm, as illustrated in Figure 5. Our previous attempt of integrating the original versions of tools was time-consuming and technically challenging because of the various discrepancies among the tools. As the tools were developed in at least three different programming languages (Java, Perl and C++), they had defined their own data formats, and adopted different offset calculation schemes (global vs. local), as mentioned before. Accordingly, we had to develop five different interfacing modules as illustrated in Figure 5a. In contrast, the integration of the respective BioC versions of all the independently developed tools (Figure 5b) was simply a matter of positioning the tools into a pipeline, as the tools could communicate seamlessly through BioC. Compared with our previous integration effort, using BioC saves >60% of lines of code in programming efforts.
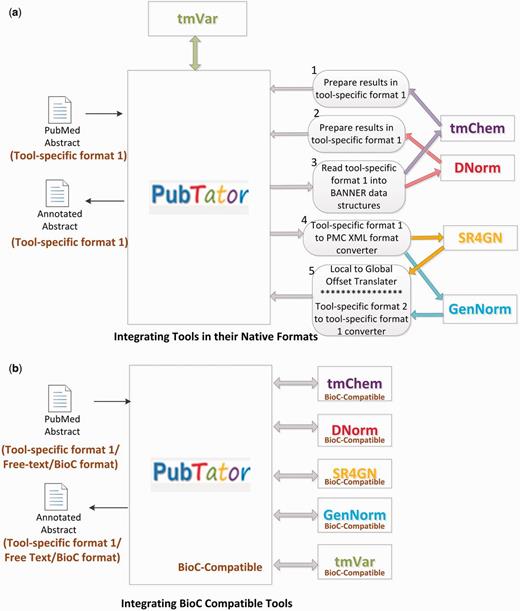
The intra-toolkit interoperability experiment. (a) Integrating tools in their native formats. (b) Integrating BIOC compatible tools.
For studying the interoperability of tmBioC with externally developed tools, we selected DNorm, the disease recognition and normalization tool from our toolkit; an externally developed tool, Abbreviations Plus Pseudo-Precision (Ab3P) (34); and a gold standard resource for disease annotation, the NCBI disease corpus (35). Ab3P is an abbreviation resolution tool that identifies <long form, short form> pairs from biomedical texts. Ab3P incorporates a pattern-matching-based algorithm implemented in C++ and has recently been equipped to process and output the BioC format. Abbreviations in biomedical domain are frequently ambiguous, leading to a number of NER errors. Hence, we added Ab3P as a preprocessing step to DNorm that replaced the short forms with long forms in the input articles. For this experiment, we trained and tested DNorm on the NCBI disease training and test corpora, recently made available in BioC, respectively. We observed 1.3% improvement in abstract-level normalization precision, over the preintegration version of DNorm. Similar to the first experiment, we were able to easily integrate and place the three BioC-compatible tools, namely DNorm, AB3P and NCBI Disease Corpus, and resources into the pipeline for supervised disease recognition and normalization system.
Discussion and conclusions
The goal of this study was to improve the interoperability of our NER tools using the recently developed BioC Family of XML formats and classes. Our toolkit consists of several competition winning, high-performing tools for concept recognition and annotation. For example, GenNorm obtained the highest performance in the BioCreative III Gene Normalization task (36), and DNorm achieved the best results the 2013 ShARe/CLEF shared task for normalizing disease names in clinical notes (1). Also, the tmVar tool for mutation recognition delivers >90% F-measure on multiple benchmarking test sets; and the PubMed-like, color-coded interface of PubTator makes it a highly usable annotation tool for human biocurators. In addition to accelerating knowledge discovery and assisting manual curation, the toolkit is capable of solving other important and challenging problems in the biomedical domain. For instance, text-mining mutation information is critical for the analysis and interpretation of sequence variations in complex diseases in the post-genomic era. Disease recognition is important for many lines of inquiry, including etiology (e.g. gene–disease relationships) and clinical aspects (e.g. diagnosis, prevention and treatment). Gene and specifies recognition could be useful for protein–protein interaction extraction.
Our experience shows that only minimal changes were required to repackage our tools with BioC and deliver the final product: tmBioC. Also, reading and writing to BioC format was fairly straightforward, as the functions and classes are already provided by the BioC authors in two widely used programming languages. For each tool, the primary developers modified their respective tools, and confirmed the simplicity and learnability of the BioC format. The primary challenge was to create the key files for the tools. However, it was a one-time effort, as all the six concept recognition and annotation tools can use a common key file for defining their BioC annotation files. The released tmBioC.key file could also evolve as a standard key file for concept recognition and annotation tasks as recommended in (37). Furthermore, we provide an easy-to-use format converter that facilitates automatic conversion among three data formats including BioC. We also provide a full-text corpus of GO annotations in BioC, which can be used to train other NER tools. All our tools are freely available and ready to be reused by a wider community of researchers in text mining, bioinformatics and biocuration communities.
Through this study, we promote the interoperability of our tools, not only with each other, but also with the tools and datasets developed by several other groups worldwide. The tools, although developed in different programming languages and following different data representation schemes, are now capable of sharing their inputs/outputs with each other, without any additional programming efforts. We confirmed this by equipping the BioC version of PubTator annotation tool with five other tools from our BioC compatible toolkit. Our tools in BioC can also interact with other state-of-the-art tools to build much more powerful applications. We confirm this by combining one of our tools in BioC with a high-precision abbreviation resolution tool in BioC, and a human-annotated disease corpus in BioC.
Our interoperability experiments only present a glimpse of the powerful applications that could be created by synergistically combining multiple tools. For example, a modular text-mining pipeline of various BioC compatible tools and corpora for NER, normalization, annotation and relationship extraction, could be developed to build sophisticated systems, e.g. an integrative disease-centered system connecting the biological and clinical aspects, providing information from causes (gene–mutation–disease relationship) to treatment (drug–disease relationships) of diseases by mining/annotating unstructured (biomedical literature, clinical notes, etc.) and structured resources (datasets released by organizations and research groups). In the future, we anticipate much broader usage of these tools, as further efforts are invested in publicizing BioC.
Acknowledgements
The authors would like to thank Don Comeau, Rezarta Dogan and John Wilbur for their discussion and help with the BioC tools, and in particular, their help on preparing the PMC articles in BioC XML format for the BioCreative IV GO task.
Funding
This research was supported by the Intramural Research Program of the NIH, National Library of Medicine.
Conflict of interest. None declared.
References
Author notes
Citation details: Khare,R., Wei,C.-H., Mao,Y. et al. tmBioC: improving interoperability of text-mining tools with BioC. Database (2014) Vol. 2014: article ID bau073; doi:10.1093/database/bau073.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}