





Abstract
Understanding prokaryotic transformation of recalcitrant pollutants and the in-situ metabolic nets require the integration of massive amounts of biological data. Decades of biochemical studies together with novel next-generation sequencing data have exponentially increased information on aerobic aromatic degradation pathways. However, the majority of protein sequences in public databases have not been experimentally characterized and homology-based methods are still the most routinely used approach to assign protein function, allowing the propagation of misannotations. AromaDeg is a web-based resource targeting aerobic degradation of aromatics that comprises recently updated (September 2013) and manually curated databases constructed based on a phylogenomic approach. Grounded in phylogenetic analyses of protein sequences of key catabolic protein families and of proteins of documented function, AromaDeg allows query and data mining of novel genomic, metagenomic or metatranscriptomic data sets. Essentially, each query sequence that match a given protein family of AromaDeg is associated to a specific cluster of a given phylogenetic tree and further function annotation and/or substrate specificity may be inferred from the neighboring cluster members with experimentally validated function. This allows a detailed characterization of individual protein superfamilies as well as high-throughput functional classifications. Thus, AromaDeg addresses the deficiencies of homology-based protein function prediction, combining phylogenetic tree construction and integration of experimental data to obtain more accurate annotations of new biological data related to aerobic aromatic biodegradation pathways. We pursue in future the expansion of AromaDeg to other enzyme families involved in aromatic degradation and its regular update.
Database URL:http://aromadeg.siona.helmholtz-hzi.de
Introduction
Aromatic hydrocarbon: the value of biodegradation
Microorganisms, mostly bacteria, play an important role in the cleanup of contaminated sites as they have acquired the ability to degrade an impressive variety of such aromatic hydrocarbon structures, using them as carbon and energy sources (1). Accordingly, the knowledge on anaerobic bacterial degradation of aromatics is constantly growing (2). However, compared with the anaerobic degradation, aerobic bacterial degradation of aromatic compounds has been much more intensively studied (1). The activation of the aromatic ring commonly proceeds by Rieske non-heme iron oxygenases (3), flavoprotein monooxygenases (4) or soluble diiron monooxygenases (5). Alternatively, activation is mediated by CoA ligases and the formed CoA derivatives are subject to oxygenations. The further aerobic degradation of di- or trihydroxylated intermediates can be catalyzed by either intradiol or extradiol dioxygenases. Contrarly to the superfamily of intradiol dioxygenases, in which all described members belong to the same superfamily, the enzymes reported to be involved in the extradiol ring cleavage of hydroxylated aromatics can be categorized in three different superfamilies: type I extradiol dioxygenases (e.g. catechol 2,3-dioxygenases), which belong to the vicinal oxygen chelate superfamily (6), type II or LigB superfamily extradiol dioxygenases which comprises, among others, protocatechuate 4,5-dioxygenases (7) and type III enzymes such as gentisate dioxygenases, comprising enzymes of the cupin superfamily (8).
Genomic data resources: living on a log scale
Alongside with the advent and fast upgrading of next-generation sequencing technologies, metagenomics and genome-wide studies, there was an exponential increase in nucleotide and amino acid sequence data (9). However, despite this overall increasing knowledge, also in aerobic aromatic degradation pathways, it is a matter of fact that this information is not clearly structured. The majority of protein sequences in public databases have not been experimentally characterized and homology-based methods are the most routinely used approach to assign and annotate protein function in sequenced genomes and metagenomes. However, the lack of a relatively high sequence identity dismisses an accurate functional assignment and leads to a large number of cumulative homology-based misannotations, which can spread through functional databases (10). In fact, misannotation in enzyme superfamilies containing multiple families that catalyze different reactions is indeed a larger problem of public databases that has been recognized (11) and protein data associated with the aromatic catabolic routes are also significantly affected by this setback (12). Taking into consideration that one crucial goal of environmental biotechnologies is to better understand the potential of microbial communities for the degradation of aromatic pollutants and to explore technologies to restore polluted environments including a systems biology approach, it is crucial to have in hand curated databases, including catabolic key proteins.
Currently available biodegradation databases
Decades of biochemical studies have produced a considerable wealth of knowledge on biodegradation, and this has started to be categorized and stored in structured databases, which have been valuable for managing biodegradation-related data. For instance, the Biodegrative Strain Database (13) lists degradative bacteria and the hazardous substrates they degrade, including the corresponding literature citations, relevant patents and links to additional biological or chemical data. The University of Minnesota Biocatalysis/Biodegradation Database (UM-BBD) (14) aims to predict plausible pathways for degradation of organic compounds based on known reactions of described microorganisms. However, a careful curation is missing in some of the pathways, such as that for 2,4-dichlorophenoxyacetate, where a 2-chlorodienelactone isomerase is still proposed to be involved (15), a reaction already refuted in 1990 (16), and important pathways are missing such as the mineralization of chlorobenzene via extradiol cleavage of intermediary 3-chlorocatechol (17). Metarouter (18) is focused on the biochemical aspects of biodegradation and allows the analysis of valuable features such as the query of pathways or networks through prediction of chemical biodegradability. However, the absence of information at the sequence level of proteins limits its use for the analysis and annotation of omic data. FunGene (http://fungene.cme.msu.edu/) (19) gives sequence information on some key proteins of biodegradation including direct links to references, however, the database takes over automatic annotations previously given and, thus, does not shed light on probable misannotations. Also Metarouter and other databases, such as the Database of Biodegradative Oxygenases—OxDBase (20) and the web-based server PathPred (21), link the entries to external databases which often direct to proteins electronically annotated in the course of complete genome sequencing projects. As an alternative, Bionemo (22) provides an update to UM-BBD, in which accurate associations between proteins and reactions are based on customized database searches, extensive literature mining and manual curation. Bionemo combines metabolic, genetic and regulatory information but unfortunately its web interface is not updated since 2008.
So far, there is no single resource that provides a direct query based on protein sequence information and data mining through a phylogenomic approach. We assumed that a web interface based on a database that consolidates all these gaps would be an invaluable tool for academic and industrial researchers as well as environmental engineers.
Construction and content
General concept: a phylogenomic approach
One of the fundamental paradigms in computational biology is functional prediction by homology (23). Generally, these predictions do not take in account that evolution not only conserves function, but it also generates new functions where the basic biochemical mechanism may be conserved, while the substrate or ligand specificity changes.
Phylogenomic analysis—combining phylogenetic tree construction and the integration of experimental data has been proposed to address these errors and improve functional classification accuracy (23). Molecular phylogenetic analysis has been used for decades for the elucidation of species relationships (24), and the importance of such analysis became more evident once Eisen (25) showed how phylogenomic analysis addresses the deficiencies of function prediction by homology and improves the accuracy (26). Since then, phylogenomic inference of protein molecular function has been applied to the detailed analyses of individual protein families (27), in comparative genomics (28) or for whole-genome analysis (29).
As previous studies have shown that a phylogenomic approach to protein functional classification results in fewer false-positive results, when compared with the pairwise methods of functional classification (26, 30) some databases such as the Lipase Engineering Databases (31), the database of epoxide hydrolases and haloalkane dehalogenases (32) or the carbohydrate-active enzymes database (CAZy) (33) have recognized and explored the relevance of phylogenomics to classify enzymes into subfamilies and to clarify the relationship between protein sequence, structure and substrate specificity.
Also the PFAM database aims to facilitate functional annotation, which here is based on domain assignments (34). This database consists of protein domain families, which are automatically classified on the basis of sequence similarities, where composition and size of the PFAM families vary significantly (34). In case of Rieske non-heme iron oxygenases, as an example, pfam00848 comprises all alpha-subunits discussed here and does not give fine-tuned information. More fine-tuned information can be obtained from the conserved domain database (CDD) which provides annotation of protein sequences with the location of conserved domain footprints (35). Actually CDD maintains an active curation effort that aims at providing classifications for major and well-characterized protein domain families, for example cd03469, the Rieske non-heme iron oxygenase family (35). However, the phylogenomic approach described here is useful for an even more detailed analysis of individual protein families crucial for the degradation of aromatics, as well as for a high-throughput functional classification at a genomic scale.
Thus, to fulfill the existing gap in the available biodegradation databases, we report here an up-to-date and manually curated database with an associated query system that exploits a phylogenomic analysis of aerobic degradation of aromatic compounds—AromaDeg. Through phylogenetic tree construction and integration of experimental data, AromaDeg addresses systematic errors produced by standard methods of protein function prediction and improves the accuracy of functional classification of key proteins of aromatic degradation.
Contents and curation
In aerobic aromatic degradation, a broad range of peripheral reactions transforms a huge variety of compounds to a restricted set of central intermediates, which are subject to ring-cleavage and subsequent funnelling into the Krebs cycle. Altogether, the catalog of oxygenases involved in activation and cleavage of the aromatic ring is extensive and phylogenetically diverse, including several different families that have been recently compiled for phylogenomic studies (12).
To avoid misleading annotations and to survey the metabolic potential of aerobic microbial communities, we developed and manually curated a database (with a total of 3605 protein sequences)—AromaDeg, focused on the following key catabolic enzymes for aromatic degradation: α-subunits of Rieske non-heme iron oxygenases, extradiol dioxygenases of the vicinal chelate superfamily, extradiol dioxygenases of the LigB superfamily and extradiol dioxygenases of the cupin superfamily (Figure 1).

Aerobic metabolism of selected aromatics via di- or trihydroxylated intermediates. Reactions catalyzed by Rieske non-heme iron oxygenases are indicated by R, those catalyzed by extradiol dioxygenases of the vicinal chelate superfamily by an E, those catalyzed by enzymes of the LigB superfamily by an L and those catalyzed by enzymes of the cupin superfamily by a C. Ring-cleavage products are channeled to the Krebs cycle via central reactions.
Rieske non-heme iron oxygenases
The Rieske non-heme iron oxygenases are one of the key enzymes important for aerobic activation and thus degradation of aromatics such as benzoate, benzene, toluene, phthalate, naphthalene or biphenyl (3) (see Figure 1, reactions indicated by R). Members of this superfamily also catalyze monooxygenations, such as salicylate 1- or salicylate 5-hydroxylases or demethylations, such as vanillate O-demethylases. It is known that the catalytic components (α-subunits) of all multicomponent Rieske non-heme iron oxygenases are related to each other and several publications attempted their classification. Nakatsu et al. (36) pointed out that all Rieske non-heme iron oxygenases devoid of β-subunits (e.g. phthalate 4,5-dioxygenase, vanillate demethylase and carbazol dioxygenase) are related to each other and form a distinct lineage of the superfamily (termed the phthalate oxygenase lineage) and Werlen et al. (37) identified four families (naphthalene, toluene/benzene, biphenyl and benzoate/toluate) in a second lineage. In general, the clustering of oxygenases into families correlates with the native substrates oxidized by their members (3). Previous phylogenomic analyses of the α-subunits of Rieske non-heme iron oxygenases (12) also identified two distinct lineages that contain proteins of validated function and correspond to the two mentioned lineages. Following this categorization, we updated these two lineages with sequence data retrieved from the NCBI website, as available in September 2013. This update was performed, as described below in the chapter ‘Building phylogenetic trees and clusters.’ Out of the phthalate oxygenase lineage, only enzymes related to those of documented function in the activation of the aromatic ring were further considered for the database and the second lineage can now be differentiated into three families—biphenyl oxygenases, benzoate oxygenases and salicylate oxygenases.
Extradiol dioxygenases of the vicinal oxygen chelate superfamily
The extradiol ring-cleavage of catechol is typically catalyzed by type I extradiol dioxygenases (EXDO) of the vicinal oxygen chelate superfamily (6). The EXDO I family comprises enzymes that catalyze the dioxygenolytic ring-fission of catecholic derivatives in several bacterial mono- and polyaromatic biodegradation pathways (38) and catalyze the extradiol-cleavage of catechol, 2,3-dihydroxybiphenyl, 1,2-dihydroxynaphthalene, homoprotocatechuate, 2,3-dihydroxy-p-cumate, 2,3-dihydroxybenzoate and 7-oxo-11,12-dihydroxydehydroabietate among others (see Figure 1, reactions indicated by E).
Phylogenomic analysis of the deduced protein sequences of EXDO I proteins encoded in the genomes of bacteria having been sequenced until August 2008 showed the presence of three major evolutionary lineages (12). One of these lineages (Lineage 1) comprises nearly all EXDO I proteins of validated function. Lineage 2 contains BphC6 of Rhodococcus jostii RHA1 (GenBank: ABO34703) and other previously characterized so-called one-domain extradiol dioxygenases such as BphC2 and BphC3 from Rhodococcus globerulus P6 with reported activity against 2,3-dihydroxybiphenyl (39) (subfamily I.1 as defined by Eltis and Bolin (38)), however, most proteins of this lineage have not been characterized thus far. Lineage 3 comprises only a few validated extradiol dioxygenases such as those involved in the turnover of (chloro)benzoquinols and (chloro)hydroxybenzoquinols (LinE chlorobenzoquinol/benzoquinol 1,2-dioxygenases (40) and PcpA 2,6-dichlorobenzoquinol 1,2-dioxygenases (41)). Among the three described lineages, AromaDeg is dedicated to Lineage 1, which comprises most of the EXDO I proteins of validated function. Analysis of the evolutionary relationships among those (38) had shown that they could be differentiated into enzymes having a preference for monocyclic substrates (termed family I.2), and those with a preference for bicyclic substrates (termed family I.3) beside above-mentioned one-domain extradiol dioxygenases (termed family I.1). Since then, an enormous amount of information has been generated (42) and more recent surveys on the phylogeny of extradiol dioxygenases (12), together with the phylogenomic analysis done here, showed that families I.2 and I.3 (38) still form groups supported by high bootstrap values. In addition, Lineage I of type I extradiol dioxygenases comprises various enzymes using miscellaneous substrates such as 2,3-dihydroxybenzoate, which were separately analyzed (family EXDO, miscellaneous substrates).
Extradiol dioxygenases of the LigB superfamily
A second superfamily of extradiol dioxygenases is the LigB-type extradiol dioxygenases, members of which are well established as being responsible for the degradation of protocatechuate via the protocatechuate 4,5-dioxygenase pathway (7). Phylogenomic analysis of the deduced protein sequences of LigB-type proteins in 2008 (12) allowed the identification of two families: the protocatechuate and the homoprotocatechuate family, which could now (as in September 2013) be verified with novel proteins mainly identified from genome sequencing projects. The protocatechuate family comprises the protocatechuate 4,5-dioxygenase β-subunits, gallate dioxygenases (43), 2,3-dihydroxyphenylpropionate dioxygenases (44) and additional LigB-type enzymes that have been described to be involved in the degradation of methylgallate (45), of bi- and polycyclic aromatics (46) or of carbazol (47) (see Figure 1, reactions indicated by L).
The homoprotocatechuate family of the LigB superfamily comprises the proteobacterial homoprotocatechuate 2,3-dioxygenases as the one described in Escherichia coli (48), the aminophenol 1,6-dioxygenases (see Figure 1, reactions indicated by L) with the β-subunits containing the active site (49) and several other LigB-type extradiol dioxygenases of unknown function, mainly observed in Clostridia and Archaea.
Extradiol dioxygenases of the cupin superfamily
Several extradiol dioxygenases of aromatic degradation pathways have been described to belong to the cupin superfamily (8) sharing a common architecture and including key enzymes such as homogentisate 1,2-dioxygenases (50) and 3-hydroxyanthranilate 3,4-dioxygenase (51). Although the above-described enzymes are involved in the degradation of amino acids by various bacteria, gentisate 1,2-dioxygenase is a ring cleavage enzyme involved in the degradation of salicylate or 3-hydroxybenzoate, among other aromatics (52) and thus, has been reported to be involved in the degradation of environmental pollutants such as naphthalene or dibenzofuran (53, 54). Although certain soil organisms, particularly of the genera Pseudomonas and Ralstonia, have received some attention with regards to the gentisate pathway (52), the importance of this EXDO superfamily for bioremediation as well as its distribution in environmental samples has thus far been neglected. Thus, AromaDeg is focused on the gentisate 1,2-dioxygenase family of the cupin superfamily, including 1-hydroxy-2-naphthoate dioxygenases, reported to be involved in the degradation of polycyclic aromatics such as phenanthrene (55) (see Figure 1, reaction indicated by C).
Building phylogenetic trees and clusters
Protein sequences of validly described members of the different catabolic protein families mentioned above had been collected and used independently as seeds for searches using the BLASTP algorithm (56) against non-redundant protein and environmental sequence databases at GenBank (10) typically with a stringent E-value threshold of 1e−20. Protein sequences of representatives of these protein families were then used as seeds for GenBank searches to cover the full range of sequences currently available (by September 2013). All protein sequences were then aligned with MAFFT (57) using default values and phylogenetic trees were constructed with MEGA5 (58) using the neighbor-joining algorithm (59) with p-distance correction and pairwise deletion of gaps and missing data. A total of 100 bootstrap replications were done to test for branch robustness.
All proteins of the respective protein family were manually checked for representatives of validated function or for gene clusters encoding that protein and comprising proteins of documented function. Due to currently biased sequencing efforts the generated trees contained various branches with identical or nearly identical proteins of the same function derived from representatives of the same species. To uphold the clearness of these trees, some redundant protein sequences (typically >95–99% of sequence identity) that were obviously over-represented in number were removed. Proteins evidently outgrouping from the constructed phylogenetic trees and belonging to other protein families were eliminated. Phylogenetic trees were inspected for evident branches supported by bootstrap analysis and for proteins of documented function as described above. Clusters were defined as branches supported by bootstrap analysis and a probable function was assigned to its members, if the cluster contains members of validated function.
As an example, the salicylate family of Rieske non-heme iron oxygenases (see also Pfam domain CD0880) of AromaDeg shown in Figure 2 contains 137 protein sequences that were sorted into 18 clusters supported by high bootstrap values and comprises enzymes capable of transforming ortho- and para-substituted benzoates. The phylogenetic tree constructed using the maximum-likelihood algorithm showed an identical topology, however, this method suffered from a low speed.
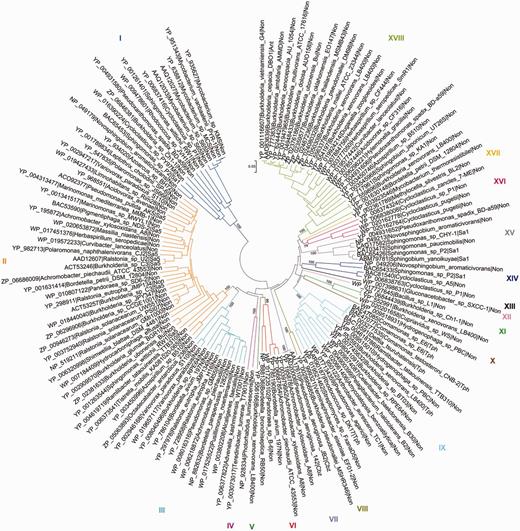
Evolutionary relationships of α-subunits of the salicylate family of Rieske non-heme iron oxygenases. Protein sequences were aligned with MAFFT and the phylogenetic tree was constructed with MEGA5 using the neighbor-joining algorithm with p-distance correction and pairwise deletion of gaps and missing data. A total of 100 bootstrap replications were done to test for branch robustness (bootstrap values are shown adjacent to each cluster node) and redundant protein sequences (>95–99% of sequence identity) were removed. According to the documented substrate specificity of representative members they can be clustered as follows: Clusters I, XIV and XV—salicylate 1-hydroxylases; Cluster II—salicylate 5-hydroxylases; Clusters III, IV, V and VI—Rieske oxygenases related to salicylate 5-hydroxylases; Cluster VII—chlorobenzoate dioxygenases; Clusters VIII, X, XI and XII—Rieske oxygenases related to terephthalate dioxygenases; Cluster IX—terephthalate dioxygenases; Cluster XVI—probable salicylate 1-hydroxylases; Cluster XVII—anthranilate dioxygenases of Burkholderia and some other organisms. Further information about each cluster is included in Table 1.
All proteins previously characterized as anthranilate, 2-chlorobenzoate or terephthalate dioxygenases group together in one cluster each (Clusters XVII, VII and IX, respectively), where it can be assumed that all proteins of the respective cluster perform the same reaction (Table 1). Similarly, phylogenomic analysis revealed that all proteins characterized thus far as salicylate 5-hydroxylases group together in Cluster II of the salicylate family of Rieske non-heme iron oxygenases whereas salicylate 1-hydroxylases could be localized in three distinct clusters (Figure 2 and Table 1).
Phylogenomic clusters of α-subunits of the salicylate family of Rieske non-heme iron oxygenases
| Clusters | Representative sequence | Annotation | Substrate | Abbreviation | Pubmed id |
|---|---|---|---|---|---|
| I | BAC65453 Sphingomonas sp. P2 | Salicylate 1-hydroxylases | Salicylate | Sa1 | 12565867 |
| II | AAD12607 Ralstonia sp. U2 | Salicylate 5-hydroxylases | Salicylate | Sa5 | 957320722936930164616532326649821369832 |
| III | ZP_06687231 Achromobacter piechaudii | Rieske oxygenases related to salicylate 5-hydroxylases | Unknown | Non | |
| IV | NP_928334 Photorhabdus luminescens | Rieske oxygenases related to salicylate 5-hydroxylases | Unknown | Non | |
| V | YP_556166 Burkholderia xenovorans LB400 | Rieske oxygenases related to salicylate 5-hydroxylases | Unknown | Non | |
| VI | ZP_06687854 Achromobacter piechaudii | Rieske oxygenases related to salicylate 5-hydroxylases | Unknown | Non | |
| VII | AAL17610 Pseudomonas aeruginosa JB2 | Chlorobenzoate dioxygenases | Chlorobenzoate | Cbz | 1172291910224014 |
| VIII | YP_999321 Verminephrobacter eiseniae | Rieske oxygenases related to terephthalate dioxygenases | Unknown | Non | |
| IX | BAE47077 Comamonas sp. E6 | Terephthalate dioxygenases | Terephthalate | Tph | 165176281267068919734336165176282288986216181748 |
| X | WP_009518931 Hydrogenophaga sp. PBC | Rieske oxygenases related to terephthalate dioxygenases | Unknown | Non | |
| XI | YP_552991 Burkholderia xenovorans LB400 | Rieske oxygenases related to terephthalate dioxygenases | Unknown | Non | |
| XII | WP_017727645 Bacillus sp. L1 | Rieske oxygenases related to terephthalate dioxygenases | Unknown | Non | |
| XIII | WP_007398631 Gluconacetobacter sp. SXCC-1 | Rieske oxygenases related to salicylate 1-hydroxylases | Unknown | Non | |
| XIV | BAC65433 Sphingomonas sp. P2 | Salicylate 1-hydroxylases | Salicylate | Sa1 | 12565867 |
| XV | BAC65426 Sphingomonas sp. P2 | Salicylate 1-hydroxylases | Salicylate | Sa1 | 125658671564939715528538 |
| XVI | YP_008374324 Cycloclasticus zancles 7 ME | Probable salicylate 1-hydroxylases | Probably salicylate | Non | |
| XVII | YP_002361789 Methylocella silvestris BL2 | Probable salicylate 1-hydroxylases | Probably salicylate | Non | |
| XVIII | AAO83639 Burkholderia cepacia | Anthranilate dioxygenases of Burkholderia and some other organisms | Anthranilate | Ant | 13129960 |
Notes. List of the three- or four-letter code indicate the experimentally validated function and/or substrate: Ant, anthranilate; Cbz, chlorobenzoate; Sa1, salicylate (salicylate 1-hydroxylases); Sa5, salicylate (salicylate 5-hydroxylases); Tph, terephthalate.
All other nine phylogenetic trees—namely, those showing the α-subunits of the phthalate, biphenyl and benzoate family of Rieske non-heme iron oxygenases; those showing families I.2, I.3 (38) and a third family of extradiol dioxygenases of the vicinal oxygen chelate superfamily; those showing the homoprotocatechuate and protocatechuate families of the LigB superfamily of extradiol dioxygenases and a tree showing the gentisate family of the cupin extradiol dioxygenases—and the cluster descriptions of the AromaDeg database are summarized and available in the Supplementary Material (Supplementary Figures S1–S9 and Supplementary Tables S1–S9).
The usefulness of the database
Blast searches (http://blast.ncbi.nlm.nih.gov/Blast.cgi) using proteins closely related to those of validated function often show highest similarity to enzymes with a proper annotation. As an example, high similarity of the protein with the GenBank accession number YP_004931652 from Pseudoxanthomonas spadix BD-a59 (E-values down to 0) was observed with enzymes annotated as salicylate 5-hydroxylases, however, also with chlorobenzoate dioxygenases such as the enzyme indentified from Pseudomonas aeruginosa 142 (GenBank accession number AAD20006, E-value 10−158) (60), whereas phylogenetic analysis was capable to clearly separate salicylate 5-hydroxylases and chlorobenzoate dioxygenases (see Figure 1, Clusters II and VII). More importantly, enzymes showing highest similarity to Cluster III enzymes, such as YP_002946168 from Variovorax paradoxus S110 (E-value of 0), are predominantly annotated as biphenyl 2,3-dioxygenases. The most similar enzymes of validated function are, however, salicylate 5-hydroxylases (e.g. salicylate 5-hydroxylase ACT53246 from Burkholderia sp. B2 (E = 2 × 10−119)) (61) and salicylate 1-hydroxylases (E = 6 × 10−86) (62). Our analysis (see Figure 1, Cluster III) clearly shows that cluster III enzymes among enzymes of validated function are most closely related to salicylate 5-hydroxylases, however representatives of this cluster need to be functionally characterized to allow any clear functional assignment.
It can, thus, be summarized that the assignment of probable functions to novel proteins involved in aromatic degradation simply based on similarity using best hits leads to functional misannotations as phylogenetically different enzymes may show equal similarities to the query enzyme. Moreover, a significant amount of misannotations is already available and landmarks of proteins with validated function are hidden behind the massive amount of information available. All these problems may be overcome by the use of AromaDeg.
The query system: a public and user-friendly web interface
To build up a searchable database with a user-friendly web interface we standardized the data source files by collecting all the compiled protein sequences in a multi-fasta format with headers composed of the protein id (accession number according to GenBank (http://www.ncbi.nlm.nih.gov)), the organism that harbors the given protein and a three- or four-letter code that describes the experimentally validated function and/or substrate. All enzymes that do not have a validated function contain the three-letter code ‘Non’ in their header.
All sequences belonging to each one of the trees are made available as a single file comprising the curated information and the complete sequence set unaligned, or aligned by MAFFT (57). All mentioned source files can be retrieved online, in the download section of the website (see link below).
The AromaDeg database can be queried at http://aro madeg.siona.helmholtz-hzi.de and in contrast to most biodegradation-related databases, it does not require a query based on previous knowledge (name of the organism, compound or enzyme) but simply a protein sequence which can be particularly advantageous when mining in novel genomes or high-throughput metagenomic or metatranscriptomic data sets. The web interface allows the query of source files up to 20 Mb. Once submitted, the retrieved information is, in a first step, compared with the entire database using the blastp program (56) with adjustable parameters. In a second step, each query sequence that matches a given protein family of the AromaDeg is aligned with the sequences constituting that protein family using MAFFT (57) with parameters ‘—globalpair—retree 300—maxiterate 300’. The multiple alignment is then used to build a phylogenetic tree using the neighbor-joining algorithm and the Ka/Ks model (63), with bootstrap values calculated to provide branch support (from 25 iterations) as implemented in the Seaview program (64). The tree file produced in Newick format is then used by the Newick Utilities program suite (65) to generate SVG (Support Vector Graphics) images of the final tree and independent clusters, which include the query of the user. Based on this result, it is possible to identify the nearest cluster the candidate sequence belongs to, and thus, obtain information on the probable protein function, substrate specificity or taxonomic classification. For this purpose, manually compiled and curated information about the members of the same cluster is available in the three- or four-letter code of the protein headers (see Figure 2 and Supplementary Figures S1–S9) and provided in the corresponding table (see Table 1 and Supplementary Tables S1–S9). Upon a successful run, the website sends an email to the user with a link where the above-mentioned result files are available for download. More details about the usage and output of AromaDeg can be retrieved online in the Extended Manual available for download.
Given the multiplicity of options, AromaDeg allows a flexible use of the manually curated biological data, ranging from an automatized pipeline where the final phylogenetic trees including the queries can be retrieved as output, to a more independent option in which the user can download the curated source files (see download section of the web interface) and perform the phylogenetic analysis using other computational tools.
Applications and future directions
Resources such as repositories of sequenced genomes and all subject-specific databases are increasingly faced with the challenge of ensuring data accuracy and efficient management and curation. However, these efforts are hampered by factors such as the reliability of curation and the lack of incentives for researchers to contribute, among others. ‘To date, not much of the research community is rolling up its sleeves to annotate’ (9). We wanted to address this issue with AromaDeg, as an effort to compile biological data related to the bacterial aerobic aromatic degradation and initiate a reliable annotation. The compilation of manually curated data has a wide range of applications in the fields of molecular biology and genomics. As a specialized database in aerobic aromatic degradation, it is not only a better-suited tool than non-specialized databases for improving the design of PCR primers and probes targeting a cluster of catabolic enzymes of a given protein family (66, 67), but also specifically for accurate annotation of genomes and high-throughput data generated by different omics approaches. A preliminary version of the biological data compilation of AromaDeg has been efficiently applied for a knowledge-based design of probes for functional gene arrays covering the diversity of aromatic degradation reactions (42) and the annotation of metagenomic sequences (68).
These successful applications have encouraged the recent update of the database and the development of a user-friendly web interface to open the database to the public and to make it suitable for high-throughput applications. To the best of our knowledge, this is the only resource available that, in a fast and precise manner, combines phylogenetic analysis and a fine level of accurate annotation of new biological data related to aerobic aromatic biodegradation pathways.
We aim in future to expand the database to other enzyme families involved in aromatic degradation, including anaerobic key enzymes and to update regularly the already described enzyme families. An upgrade of the database by enzymes of validated function, which may have been overseen in the database construction, is for sure welcome. We therefore expect this tool to be used in a broad spectrum of scientific and applied research. Furthermore, we strongly believe in the robustness of this approach for reliable function prediction and we envision its use in wide range of fields, other than biodegradation, to hopefully allow researchers to dive deeper in the functional properties of enzymes.
Accessibility
AromaDeg is freely available online for educational and research purposes by non-profit institutions at http://aromadeg.siona.helmholtz-hzi.de. All sequences described in this article can be downloaded from that site.
Acknowledgments
M.D. acknowledges the support from the Helmholtz International Graduate School for Infection Research (HZI Graduate School).
Funding
European Community Project MAGICPAH (FP7-KBBE-2009-245226).
Conflict of interest. None declared.
References
Author notes
†These authors contributed equally to this work.
§Present address: Ramiro Vilchez-Vargas, Laboratory of Microbial Ecology and Technology—LabMET, Ghent University, Coupure Links 653, 9000 Gent, Belgium.
Citation details: Duarte,M., Jauregui,R., Vilchez-Vargas,R., et al. AromaDeg, a novel database for phylogenomics of aerobic bacterial degradation of aromatics. Database (2014) Vol. 2014: article ID bau118; doi:10.1093/database/bau118

{kind=link}
{kind=link}