





Abstract
miRò is a web-based knowledge base that provides users with miRNA–phenotype associations in humans. It integrates data from various online sources, such as databases of miRNAs, ontologies, diseases and targets, into a unified database equipped with an intuitive and flexible query interface and data mining facilities. The main goal of miRò is the establishment of a knowledge base which allows non-trivial analysis through sophisticated mining techniques and the introduction of a new layer of associations between genes and phenotypes inferred based on miRNAs annotations. Furthermore, a specificity function applied to validated data highlights the most significant associations. The miRò web site is available at: http://ferrolab.dmi.unict.it/miro.
Database URL:http://ferrolab.dmi.unict.it/miro
Introduction
Post Transcriptional Gene Silencing (PTGS) is a highly conserved mechanism of gene expression regulation and microRNAs (miRNAs) are its main actors. These little RNA molecules are able to bind to specific sites located in the 3′ untranslated regions (UTRs) of target transcripts, inhibiting their translation or promoting their degradation (1,2). Although much effort has demonstrated their crucial role in several physiological and pathological processes, their mechanisms of action still remain unclear.
The knockout of a single miRNA may dramatically affect the phenotype of an organism (3). Evidence suggests that a miRNA may target more than one gene, often in several sites, and that one gene may be targeted by many miRNAs acting cooperatively. Many computational predictions of miRNA targets are available on the web, but a precise association between miRNAs and phenotypes has been demonstrated only for few cases. Much more is known about genes: for example, the Gene Ontology (GO; http://www.geneontology.org/) database (4) provides annotations for processes and functions in which they are involved. Moreover, there is a vast literature concerning genetic roles in pathologies. Nonetheless, miRNAs may be annotated with information about their validated or predicted targets. For example it is known that the antiapoptotic gene BCL-2 is involved in ‘B-Cell Chronic Lymphocytic Leukemia’ (CLL) and it has been shown that low expression of miR-15a and miR-16 is related to over-expression of BCL-2 in a number of CLL patients (5). Consequently, miR-15a and miR-16 may be functionally associated with ‘apoptosis’ and ‘CLL’.
A common approach in the study of diseases or biological processes involving miRNAs, requires the extraction of data from several independent sources, such as miRNA/target prediction databases, gene functional annotations, expression profiles and biomedical literature.
Thus, there is a need for a system that integrates data from heterogeneous sources in order to build extensible and updatable knowledge bases. This system should be equipped with mining algorithms capable of inferring new knowledge.
The first system for functional annotation of miRNAs is miRGator (6), a database which integrates data from different sources, like target prediction tools and GO, and makes it available through a set of standard queries. Although miRGator represents a first attempt at the integration of such data, it has several limitations. It does not provide information about the diseases, nor does it implement customizable queries or data mining facilities.
Here we present miRò, a new system which provides users with miRNA–phenotype associations in humans. miRò is a web-based environment that allows users to perform simple searches and sophisticated data mining queries. The main goal of miRò is to provide users with powerful query tools for finding non-trivial associations among heterogeneous data and thereby to allow the identification of relationships among genes, processes, functions and diseases at the miRNA level. Finally, the data mining module includes a specificity function allowing selection of the most significant associations among validated data.
Description of the system
Data Integration
The miRò web site integrates data from different sources, as shown in Figure 1. miRNAs are annotated with information about their precursor and mature sequences coming from miRBase (http://microrna.sanger.ac.uk/sequences/) (7), and with expression profiles obtained from the Mammalian microRNA Atlas (8). The miRNA Atlas contains expression patterns of pre-miRNAs and mature miRNAs in several kinds of tissues, both normal and malignant. miRNAs are also associated to GO terms and diseases through their targets: each miRNA inherits all the annotations of its target genes. Experimentally supported miRNA/target pairs come from miRecords (http://mirecords.umn.edu/miRecords/) (9). The predicted targets are taken from the web sites of TargetScan (http://www.targetscan.org/) (10), PicTar (http://pictar.mdc-berlin.de/) (11) and miRanda (http://www.microrna.org/) (12). The target genes records are enriched with general information such as genomic context and transcript-related data coming from the NCBI Gene and Nucleotide Databases (http://www.ncbi.nlm.nih.gov/). The ontological terms with which the target genes are annotated (processes and functions) are obtained from the GO Database. Finally, the gene–disease relations come from the Genetic Association Database (GAD; http://geneticassociationdb.nih.gov/) (13), which is a database of human genetic association studies of complex diseases and disorders.
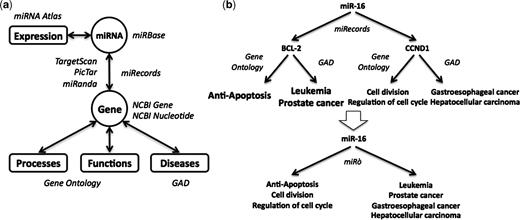
The miRò knowledge base schema. (a) miRNAs are annotated with their features coming from miRBase and their expression profiles coming from the miRNA Atlas. They are linked to processes, functions and diseases through their predicted (by TargetScan, PicTar and miRanda) or validated target genes (miRecords). (b) In this case, miR-16 has two validated targets, BCL2 and CCND1, among others. These genes are annotated with GO terms and diseases, thus miR-16 inherits these annotations.
All the data are collected and maintained up-to-date in a MySQL database. In particular, the most relevant data about the miRNAs and the target genes, such as the genomic contexts and the sequences, are stored in the database for an immediate availability, while links to the original sources are provided for more detailed information. The data are retrieved from the source websites as flat files except for GO, which is provided as a mySQL db dump and the miRNA Atlas, which is given as a collection of Excel spreadsheets. Initially, miRNA information from the miRBase files is stored. Then, all the target prediction data are screened and stored together with information on the target genes, retrieved from the NCBI Gene files. Gene aliases are also stored, in order to facilitate the subsequent integrations. In the prediction files, the genes are identified by their NCBI IDs, while the miRNAs are identified either by their accessions (miRanda) or their IDs (TargetScan and PicTar). The genes are then annotated with their GO terms, coming from the GO Database, and their associated diseases, retrieved from GAD. Since in both cases the genes are identified by their names, the gene aliases are often used in this step, in order to correctly identify all of them. Finally, miRNA expression profiles are integrated. For each miRNA, identified by its ID, all the expressing tissues, together with the expression values, are stored. The percentage distribution of miRNA clones in the tissues is also computed and then stored for fast retrieval.
Inconsistencies in data integration are prevented by a semi-automatic process: the system automatically detects and reports in a logfile any mismatches, which are then resolved manually. For example, all the miRNA names in the prediction files (PicTar and TargetScan) which can not be found in the miRBase data (e.g. their IDs have been changed), are reported in the logfile. The automatic procedure also retrieves all the similar names in the database. These are then manually screened, sometimes using also sequence information, in order to find the correct ones.
The system also automatically checks for new releases of the source databases every three months and performs an update if needed.
The miRò web interface
The miRò web interface allows the user to perform four different types of queries. A simple search is used to get information about a single object, which can be a miRNA, a gene, a process, a function, a disease or a tissue. For example, it is possible to specify a miRNA or to choose one from the complete miRNA catalog to get the list of all the diseases and GO Terms (Processes and functions) which can be associated to that miRNA through its targets. The results are ordered by the number of tools which predict the corresponding miRNA–target pairs and the experimentally supported associations are always given first. Using the ‘AND’ constraint enables users to select only the terms associated to the targets predicted by all the selected tools. This may help to identify the most strongly supported associations, and reduce the falsely predicted associations.
Similarly, the user can search for all the miRNAs associated to a certain gene, disease, process or function and obtain a list of all the miRNAs expressed in a certain tissue with their expression levels.
A customized search also allows users to extend the knowledge base by a personal set of miRNA–target pairs. These pairs will be temporarily stored and used in all the session queries. This feature may be helpful in testing new miRNA–target data.
The advanced search form can be used to perform more sophisticated queries. The user chooses a ‘subject’ among miRNA, gene, disease, process and function, then specifies a list of constraints that the subject must satisfy.
For example, it is to possible to ask miRò to show all the miRNAs (the subject is ‘miRNA’) which are associated to ‘heart failure’, ‘RNA binding’ and ‘apoptosis’, but not to ‘congenital heart diseases’. The user may also choose the sources of the miRNA–target pairs. This allows to tighten or relax the query conditions, in order to get a smaller or a larger output, respectively. The system will show the list of all the miRNAs which satisfy these constraints, with details about the involved targets (Figure 2). This query tool links objects through miRNA-based associations. For example, a disease d and a process p which are not linked through any common gene might be associated through a miRNA which regulates a gene gd, involved in d, and a gene gp, involved in p. This introduces a new layer of associations between genes and processes inferred based on miRNAs annotations. These associations are given in the Advanced Search results pages, when the subject of the query is a GO term or a disease.
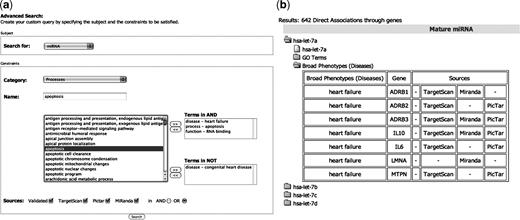
An example of Advanced Query execution. (a) The advanced search form with the selected constraints: miRNAs involved in heart failure, apoptosis and RNA binding, but not in Congenital heart disease will be returned. (b) A subset of the corresponding results. For example, the miRNA let-7a satisfies the specified requirements. In particular it is predicted to bind seven genes, which are involved in the heart failure disease. The details about the other constraints (apoptosis and RNA binding) are shown in the GO Terms folder.
Data mining
Maximal frequent item sets of miRNAs
Since miRNAs are associated to GO terms and diseases, they can be clustered according to their common terms, i.e. miRNAs which are associated to the same set of terms are grouped together. miRò is equipped with a data mining module, based on a maximal frequent item set computation (14,15), which allows users to query the database and extract non-trivial subsets of miRNAs sharing some features. The analysis is performed using different support thresholds: a high threshold allows to obtain a small number of miRNA subsets associated to a great number of terms, while a low threshold gives more subsets associated to fewer terms. All the subsets have been pre-computed off-line on the dataset of the validated miRNA–target pairs.
In the miRò interface, the user may choose up to n miRNAs together with an association criteria (i.e. process or disease). The system will find all the subsets of the selected miRNAs and the processes or diseases which they are most closely associated to. This may suggest that a set of miRNAs acting cooperatively carry out certain biological functions, as will be shown in the next section. Moreover, miRNA–term associations are scored in order to highlight the most significant ones, as discussed in the next subsection.
Specificity of miRNA and phenotype associations
Intuitively, a gene associated with fewer processes is more focused on the ones remaining. The specificity of a miRNA for a process relies on the number of targets and their specificity to the process.
This function has been applied to the set of validated miRNA–target interactions. The subsets of frequently associated miRNAs are visualized by tables showing the miRNAs and the processes/diseases to which they are all associated, with their specificity scores. The table entries are colored based on the specificity value ranging from blue (lowest value) to red (highest value) (Figure 3).
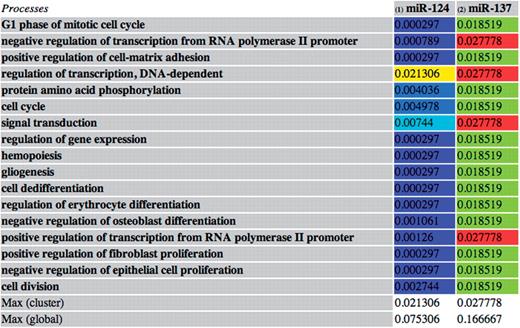
An example of an miRNA subset containing two miRNAs (miR-124 and miR-137) both involved in 17 processes. The entries contain the miRNA–process specificity scores. The entries are colored based on their value: the red entries indicate the maximum value of the subset, while the blue ones indicate the minimum values. In this case, the most relevant associations in the subset are between miR-137 and the four processes corresponding to the red entries. This may suggest a specific role of miR-137 in such processes and is due to the number of targets of the miRNA involved in such processes and to their specificity to the processes.
Use cases and validation
The system has been tested on some known cases coming from the literature. It has been able to identify miRNA–disease and miRNA–process associations previously reported, as shown in the next subsections.
The miR-17-92 cluster
The role of the miR-17-92 cluster in development and disease had been well established (16,17). The expression of these miRNAs promotes cell proliferation, suppresses apoptosis of cancer cells, and induces tumor angiogenesis. In particular they are involved in ‘lymphoma’, ‘melanoma’ and other types of cancer (‘breast’, ‘colorectal’, ‘lung’, ‘ovarian’, ‘pancreas’, ‘prostate’ and ‘stomach’). The miR-17-92 cluster also plays an essential role during normal development of the heart, lungs, and immune system.
Performing an advanced search on miRò that searches for the diseases related to subgroups of the miRNAs of the cluster, one finds that four of them (miR-17, miR-19a, miR-19b and miR-92a) are associated to those tumors together with other pathologies. Moreover, an advanced search for the processes involving the cluster returns, among others, ‘angiogenesis’, ‘apoptosis’, ‘cell cycle’, ‘cell growth’ and ‘proliferation’, ‘heart and lung development’. These processes are also linked to the diseases reported in (16,17).
Other cases
The search through the miRò Advanced Search form for the miRNAs miR-1, miR-206 and miR-133a, independently known to be involved in muscle activity (18), shows the involvement of such miRNAs in ‘muscle contraction’. Moreover, the data mining analysis detects a high correlation of miR-1 and miR-206, which are frequently associated in terms of both biological processes and pathologies.
Similarly, miR-124 and miR-137, which have been independently reported to be involved in ‘glioblastoma’ (19), are associated together to several processes, among which ‘gliogenesis’.
Validation of the specificity function
The specificity function, introduced in the previous section, aims at scoring the miRNA annotations in order to highlight the most significant ones.
Among the top ranking miRNA–disease and miRNA–process associations, there are cases which have been reported in literature, as shown in Tables 1 and 2. For example, the top scoring miRNA–disease association links miR-433 to ‘Parkinson's disease’. This result is confirmed by a study which has shown that the disruption of the miR-433-binding site of the gene FGF20, confers risk for Parkinson's disease. Indeed, the increase in translation of FGF20 is correlated with increased alpha-synuclein expression, which is known to cause Parkinson's diseases (20).
Top ranking miRNA–process associations reported in literature
| Rank | miRNA | Process | Reference |
|---|---|---|---|
| 2 | miR-212 | Cell–cell junction assembly | (27) |
| 6 | miR-224 | Apoptosis | (21) |
| 7 | miR-433 | Fibroblast growth factor receptor signaling pathway, Cell growth | (20) |
| 9 | miR-221 | Cell-cycle arrest | (28) |
| 9 | miR-222 | Cell-cycle arrest | (28) |
| 11 | miR-219-5p | Apoptosis | (29) |
Similarly, the association between miR-224 and ‘apoptosis’ is among the top ranking miRNA–process associations. This is supported by a study showing that miR-224, which is up-regulated in Hepatocellular carcinoma patients, increases apoptotic cell death by targeting the apoptosis inhibitor-5 (API-5) (21).
Conclusions
miRò is an extensible web-based system which provides users with miRNA functional annotations inferred through their validated and predicted targets. The system is conceived as an extensible and automatically updatable knowledge base with powerful data mining facilities. The main novelty of miRò is the introduction of a new layer of associations between genes and processes/diseases based on miRNAs annotations and a specificity function which allows the selection of the most significant associations among validated data.
Several new data sources will soon be integrated including pathway data, target polymorphisms and target expression levels as well as cross-species conservation data. We are also developing a new scoring function for miRNA's annotations based on the semantic associations between terms, mined from the biomedical literature.
Funding
U.S. National Science Foundation grants IIS-0414763, DBI-0445666, N2010 IOB-0519985, N2010 DBI-0519984, DBI-0421604 and MCB-0209754 (to D.S.).
Conflict of interest statement: None declared.
Acknowledgements
The authors kindly thank Chris Sander and Doron Betel for their valuable suggestions.
References
Author notes
The authors wish it to be known that, in their opinion, the sixth and seventh authors should be regarded as joint authors.

{kind=link}
{kind=link}
{kind=link}