





Abstract
A major challenge for functional and comparative genomics resource development is the extraction of data from the biomedical literature. Although text mining for biological data is an active research field, few applications have been integrated into production literature curation systems such as those of the model organism databases (MODs). Not only are most available biological natural language (bioNLP) and information retrieval and extraction solutions difficult to adapt to existing MOD curation workflows, but many also have high error rates or are unable to process documents available in those formats preferred by scientific journals.
In September 2008, Mouse Genome Informatics (MGI) at The Jackson Laboratory initiated a search for dictionary-based text mining tools that we could integrate into our biocuration workflow. MGI has rigorous document triage and annotation procedures designed to identify appropriate articles about mouse genetics and genome biology. We currently screen ∼1000 journal articles a month for Gene Ontology terms, gene mapping, gene expression, phenotype data and other key biological information. Although we do not foresee that curation tasks will ever be fully automated, we are eager to implement named entity recognition (NER) tools for gene tagging that can help streamline our curation workflow and simplify gene indexing tasks within the MGI system. Gene indexing is an MGI-specific curation function that involves identifying which mouse genes are being studied in an article, then associating the appropriate gene symbols with the article reference number in the MGI database.
Here, we discuss our search process, performance metrics and success criteria, and how we identified a short list of potential text mining tools for further evaluation. We provide an overview of our pilot projects with NCBO's Open Biomedical Annotator and Fraunhofer SCAI's ProMiner. In doing so, we prove the potential for the further incorporation of semi-automated processes into the curation of the biomedical literature.
Introduction
MGI (http://www.informatics.jax.org), the model organism database for the laboratory mouse, provides a comprehensive, integrated public information resource of Mus musculus genetics, genomics and biology (1,2). This vast catalog of integrated biological information contains extensively curated mouse data that spans from DNA sequence to disease phenotype. To collect, curate, structure and store this disparate data, MGI relies on a combination of literature curation, data loads, computational curation (evidence inferred from electronic annotation) and collaboration with other online bioinformatic resources, including SwissProt, InterPro and NCBI. More than 30 full-time curators, system administrators and support staff actively support and contribute to MGI database projects (1).
For literature curation, MGI focuses on the primary literature. MGI curators regularly review more than 160 scientific journals in electronic format (PDF or HTML) for information relevant to mouse biology. We screen more than 12 000 articles per year for potentially significant references to include in the MGI knowledge base. During primary and secondary literature selection, papers are manually selected and catalogued in a master bibliography section of the MGI database system. Selected articles are then further categorized and meticulously indexed by curators, who identify the type of mouse data contained in the article and tag articles to be indexed within the MGI database. Individual curation teams are responsible for managing Gene Ontology (GO), gene expression, sequence, mapping, phenotype and tumor data. Each team has their own methodology for indexing, which is our internal process for associating articles selected for curation to at least one entity within the MGI database. For the GO team, this entity is a gene, usually identified by a gene symbol, name, or synonym. Because gene indexing identifies papers for further curation of more detailed data that will be represented in MGI, it is a prerequisite step required for streamlining and organizing additional curation tasks. Each paper must be indexed to at least one gene entity before it enters the annotation stream. Once indexed, papers are assigned to curators for annotation according to areas of experimentation. All papers selected for indexing and curation are archived in PDF format within an internal MGI editorial database.
Defining an MGI text mining prototype project and system specifications
Although there are many areas within the MGI curatorial workflow that could potentially benefit from text mining applications, we selected gene indexing as an ideal test case for evaluating such tools to help streamline our curation procedures (see Figure 1). We index only the mouse genes that are the main topic of a review or the subject of new data, as opposed to secondary genes mentioned in the discussion section or references. In many cases, the article title and abstract clearly identify the primary genes. The exceptions—papers in which primary genes are buried in the body copy, materials and methods, or figure captions—are what make this task difficult, if not impossible, to fully automate. Because biomedical research papers tend to be littered with gene names and synonyms, some of which may be commonly used English words or acronyms, gene indexing in general can be tedious and time consuming (2–7). For MGI, it is even more challenging as mouse genes need to be distinguished from human genes of the same name and from gene mentions associated with transgenic mouse models, which are not incorporated in the GO annotations.
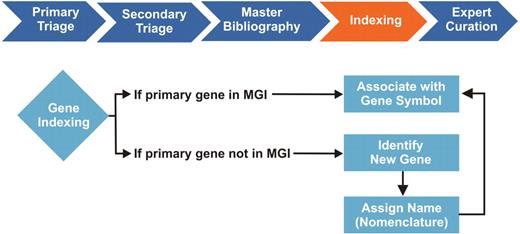
Gene Indexing in the MGI Biocuration Workflow. During primary and secondary literature selection (triage), MGI curators review scientific journals in PDF or HTML format for information relevant to the mouse model organism. We initially screen articles by searching for the terms: Mouse, Mice and Murine. Relevant articles are retrieved in PDF format, cataloged based on content, and assigned an internal reference number, which is entered into a master bibliography. Selected articles are then further categorized and indexed by curators, who identify the type of mouse data contained in the article and tag articles for indexing in the MGI database. Individual curation teams are responsible for managing GO, gene expression, sequence, mapping, phenotype and tumor biology data. Each curation team has its own methodology for indexing, which is our internal process for associating article reference numbers to at least one entity within the MGI database. For the GO curation team, this entity is a gene, typically identified by a gene symbol, name or synonym. During gene indexing, curators identify the primary mouse genes studied in the article. Mouse gene references must first be distinguished from human genes of the same name and from gene mentions associated with transgenic mouse models, which are not incorporated in the GO annotations. If an identified mouse gene is in MGI, the article reference number is associated with the gene in the database and the article enters the curation queue. If the gene is not in MGI, it is identified as a new gene, forwarded to Nomenclature for name assignment, where the new gene is added to the database. The article is then associated with the newly created gene symbol and ready for curation. The gene indexing process identifies papers for further curation of more detailed data. Each paper must be indexed to at least one gene entity before it can be assigned to curators for annotation.
On average, one part-time curator assigned to this function indexes two papers an hour, depending on the complexity of the articles (poorly written articles are more difficult and time-consuming to index). Without the aid of text mining tools, we index ∼200 papers per month. With more than 1000 articles flooding the MGI annotation pipeline each month, ∼700 of which are selected for GO, gene indexing causes a significant bottleneck for MGI curators. Our objectives as we initiated this software search were first to identify, then implement a text mining solution that could minimize this bottleneck. We plan to use the results of this prototype implementation as a guide for other potential text mining software projects within MGI.
Surveying the state of the art in biomedical text mining
Text mining and natural language processing (NLP) are far from trivial (8). The content of human language cannot be captured in precise algorithms. Consequently, most NLP systems have to perform text analysis by splitting processes into smaller sub-tasks, such as breaking text into units (a sentence, word, number, or delimiter), chunking sequences of these units into concept names and entities, annotating words in the context of the role they play in a sentence (nouns, verbs, articles, etc.), and performing syntactical parsing to analyze sentences according to basic rules of grammar (9). These tasks are further complicated by the requirements of bioNLP, which is centered around biomedical literature in which gene names and other specific biological terms can be common English words and the lack of consistency in the use of biological terminology is pervasive (3,4,6,7). In addition, most scientific journals have standardized on PDF or HTML formats that, while ideal for article dissemination, are problematic for bioNLP functions. A related problem is that PDF conversions required to produce plain text (TXT) submissions for bioNLP systems are often error prone, and special characters, symbols, text formatting and columnar text flow can be lost in translation.
Biologists and text mining system developers have been working on these types of complex problems for many years, and numerous software solutions have emerged that provide powerful and sophisticated methods of biomedical information retrieval (IR) and information extraction (IE) (5,8). Many of these programs were originally developed in response to text mining community challenges posed by the Text Retrieval Conference (TREC) and BioCreative (2,3). These highly specialized text mining applications incorporate a blend of bioNLP capabilities, complex algorithms and rules based on scientific vocabularies defined in standard dictionaries and ontologies (3–8).
We focused on identifying systems designed to perform the bioNLP subtasks most important for our gene indexing function: information extraction tools, more specifically named entity recognition (NER) software, and tools for identifying protein interactions and relations (8). After careful review of various text mining system design specifications (and the effort and expertise required to develop and maintain these systems), MGI confirmed it made more sense to ‘buy not build’ a gene entity recognition application, with the caveat that no one ‘off the shelf’ system would be perfect as delivered, nor could one tool automate all aspects of biomedical information retrieval and extraction performed at MGI.
Evaluating text mining tools for MGI gene indexing
To determine which solution was best suited to streamline the MGI gene indexing function, we approached the project much like any major software search and evaluation process (see Figure 2). We first documented our system requirements, creating a software evaluation checklist and performance metrics based on our existing triage and annotation procedures (see Table 1). These documents summarized the project objective, required text mining capabilities, desired input and output options, performance measurement guidelines and cost considerations. For information extraction systems, the gold standard performance measurement is the F-score: the harmonic mean of precision and recall, which are statistical measures closely related to specificity and sensitivity (4,5,9) (see Table 2). During our initial product evaluations, we elected to screen potential NER solutions based on published BioCreative F-scores (or comparable performance measurements for systems not participating in BioCreative challenges). In addition, we relied heavily on articles summarizing the results of BioCreative challenges and initiatives to identify systems that could best address our specific curation needs (3–5). Our search was biased toward relatively mature solutions and those available from organizations with which we could potentially collaborate.
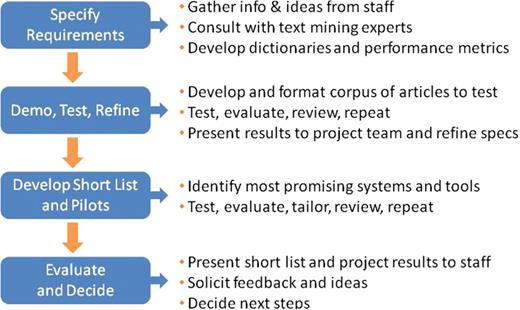
MGI software evaluation process for text mining applications. MGI applied the basic steps for managing any major software acquisition project to evaluate potential text mining applications for gene indexing.
MGI gene indexing system requirements checklist
| Evaluation criteria | Requirement objective and comments |
|---|---|
| □ Scan full text articles and perform entity recognition for mouse gene mentions (official gene symbol, name or synonym) based on a dictionary of mouse genes and human orthologs. | To streamline and optimize the gene indexing task of identifying primary genes studied in each article. |
| □ Process PDF files in batch. | To speed and simplify document processing, complement existing PDF-based literature selection and curation processes, and minimize PDF file conversion errors. |
| □ Produce reports that provide information on frequency of gene mentions by section. | To semi-automate the gene indexing task by reporting relevance scores for each gene mention detected. Relevance scores should be calculated based on frequency of gene mention occurrence, weighted by the section in which the gene entity was identified. Gene entities detected in article references, for example, have low value. |
| □ Provide visualization that highlights gene mentions in the context of the article text and original document layout. | To complement existing literature selection and curation processes. Curators are more efficient when working with tagged text in the original journal format or with clear visual cues. |
| □ Incorporate user-customizable dictionaries and ontologies to support additional curation tasks. | To adapt the tool for other potential text mining applications and curation tasks. |
| □ Achieve BioCreative F-scores of 80% or higher, or comparable performance scores. | To screen potential bioNLP tools for in-house testing and to verify that the tool will consistently identify primary genes discussed in the article. |
| □ Increase curator productivity and efficiency. | To ensure the tool makes curation tasks easier, not more difficult or more time consuming. |
Information extraction performance metrics
| Metric Term | Definition |
|---|---|
| True Positive (TP) | Number of genes detected correctly (present and marked) |
| False Positive (FP) | Number of genes detected incorrectly (marked when not present) |
| False Negative (FN) | Number of genes not detected (present, but not marked) |
| Precision (P) | |
| Recall (R) | |
| F-score |
To meet our minimum criteria, we determined that the system needed to be able to scan full-text articles for gene mentions in the form of an official gene symbol, name, or synonym. Desired features included the ability to process PDF files in batch, to produce meaningful reports that provide information on frequency of gene mentions by section, to provide visualization tools that highlight gene mentions in the context of the article text, to incorporate user-customizable dictionaries to support additional curation tasks, and to achieve BioCreative F-scores of 80% or higher. Most importantly, we wanted a tool our curators would actually use, one which made the indexing task easier, not more difficult or more time-consuming. For example, we hoped to find applications that could be integrated into our MGI editorial production system, so that curators could specify annotation parameters, upload files, and run jobs remotely from their desktop computer of choice (PC or Mac), without having switch to another program or learn commands for an unfamiliar operating environment or programming language.
Our project team met with MGI staff to gather information and ideas. We consulted with text mining experts, including bioNLP pioneer Lynette Hirschman of the MITRE Corporation and bio-ontology specialist Nigam Shah at the National Center for Biomedical Ontology (NCBO) (3,10). We used the information gleaned from these informal interviews to compile a list of text mining tools for evaluation. Our objective was not to conduct an exhaustive review of all available bioNLP packages, but rather to identify text mining solutions suitable for biocuration tasks such as ours. We then expanded and enriched this working list by incorporating the published results of BioCreative tasks and challenges for gene mention identification and gene normalization (2,5–7).
We also assembled materials and tools needed to objectively evaluate and compare different types of text mining systems. These included:
A pilot corpus of 100 articles selected from the MGI gene indexing pipeline in multiple file formats (PDF, HTML, RTF and TXT) that represented papers in which the primary genes were easy to identify from the article title or abstract as well as those that were more challenging to index,
A comprehensive dictionary of mouse gene names, human orthologs and synonyms in CSV format, and
A collection of PDF conversion utilities, such as IntraPDF and PDFTron, which could process multiple PDFs in batch, with minimal conversion errors.
After extensively testing multiple conversion utilities, we found that the quality of document conversions varies dramatically depending on the utility used, the layout of the original document, and the presence of ASCII characters and special character formatting (such as superscript and subscript). This spurred us to amend our performance measurements to note that the quality of source document (the converted PDF file) could affect the overall performance of the text mining tool.
Developing the MGI text mining application shortlist
The next step in our evaluation process was to test different systems using our pilot corpus of articles, summarize and present the results to MGI GO curation team, then refine system specifications based on their feedback. This enabled us to identify which features were most helpful to curators responsible for gene indexing, to refine our requirements checklist, and to develop a short list of text mining applications for more rigorous testing.
During the information gathering stage, we focused on two flavors of bioNLP tools: those designed to search a body of literature (typically a literature database containing only abstracts of each article) and retrieve a list of relevant articles based on user specified terms; and those designed to scan a user-specified set of full-text articles for relevant terms and concepts using dictionaries and NLP rules (8). As an example of the former, iHOP is a web service that enables users to craft a complex query to retrieve a list of articles from a literature or knowledge base, such as PubMed, using specific gene and protein related terms (11). This web service not only returns a list of sentences with the biological terms of interest highlighted, but it also enables users to build a ‘gene model’ that graphically depicts loose gene associations based on selected literature references (11,12). Other tools of this type include GoPubMed and Textpresso, both of which are particularly well suited for tailoring literature searches to include specific GO terms and phrases (13,14). These are all powerful information retrieval tools, one or more of which should be in the IR toolbox of every researcher and curator. Even though IR solutions are not well suited for automating gene indexing as implemented at MGI, we found that by evaluating and testing a broad variety of text-mining tools, we gained a better understanding of the range of open-access bioNLP systems and web-service interfaces available.
We found our needs were more closely met by NER systems designed to locate positions within the text detected as gene names (gene mention taggers) and to produce a list of unique gene identifiers for the gene and gene products tagged in the text (gene normalization) (4–6,8). We spent considerable time testing systems that scored well in BioCreative challenges for these tasks (3–5).
One example is the gene mention tagger AIIAGMT, which was developed by Cheng-Ju Kuo's lab at the Institute of Information Science, Academia Sinica, in Taiwan. It was reviewed in a BioCreative 2 challenge and placed second in the 2008 BioCreative challenge competition for gene normalization (6). An open access web tool and a BioCreative MetaServer annotation server, this system applies a modified GENIA gene mention tagger and boosts gene normalization results by applying a specialized set of approximate string matching algorithms and classifiers (5,15). It can process large blocks of plain text input or retrieve article abstracts for processing by PubMed ID (PMID). AIIAGMT also provides a nice visualization tool for output that highlights gene mentions in text (5).
Our project evaluation team found this tool easy to use and fast. In our in-house tests, it typically took <30 s to process a 3000-word block of text. According to BioCreative performance testing, it received a solid F-score of 0.75 (5) (see Table 2). Most importantly, our gene indexing staff found it helpful for scanning abstracts and sections of articles. For MGI, the primary limitation of this service was that it identified human genes only using an Entrez Gene human dictionary, and had no options for tagging entities using alternate dictionaries, such as the MGI mouse gene dictionary. Other drawbacks were related to input and output options. The AIIAGMT server can process only 3000 words in plain text format at a time. This means full text articles must be converted into text files, then broken into sections before they can be submitted to the service. AIIAGMT offered no reporting, no batch processing capabilities and it was not customizable. For our purposes, we determined it was a very good, accessible tool, suitable for occasional gene-tagging tasks, but it was not appropriate for incorporating into our production environment.
After evaluating and testing different text mining solutions, we identified two IE systems for our short list: the Open Biomedical Annotator (OBA) from NCBO and ProMiner from Fraunhofer SCAI (10,16). Although neither of these text mining tools fit our system specifications exactly, we found that each had different strengths that warranted additional testing. We established working communications with the project managers for OBA and ProMiner (Nigam Shah and Juliane Fluck, respectively).
NCBO Open Biomedical Annotator
OBA is an ontology-based system developed and maintained by NCBO at the Stanford Center for Biomedical Informatics Research (10). This open access web service is designed to annotate raw text and generate annotation reports using semantic web standards. It processes plain text submissions using a ‘concept recognition tool’ and associated dictionaries to identify relevant ontology concepts and generate direct annotations. These direct annotations are fed into semantic expansion components, which enhance the original annotations using semantics stored in one or more user-specified ontologies. OBA then produces a detailed report of the semantically expanded annotations for the text, with associated relevance scores for each recognized term and a reference to where the term was located in the text (10).
During this evaluation project, MGI worked with NCBO developers to incorporate our MGI dictionary of mouse genes and human orthologs into a pre-production version of OBA system, and to include the following OBO Foundry open biomedical ontologies, with the OBO Foundry Prefix noted in parenthesis, which we felt might be important for future MGI text mining initiatives: OBA's primary strengths are that it was designed to be customized and tailored by different users based on their annotation requirements, and it is fully supported by NCBO. Although OBA was in beta release and the development server frequently unavailable when we first began testing it, the system matured and became more useful over the course of our pilot project. It is now publically available on the NCBO BioPortal.
Human Disease (DOID);
Human Developmental Anatomy (EHDA and EHDAA);
Gene Ontology (GO);
Mouse Gross Anatomy and Development (EMAP);
Mouse Pathology (MPATH);
Mammalian Phenotype (MP).
From MGI's perspective, one of the biggest limitations of this (and other text mining systems that require plain text input) was that it required error-prone conversion of PDFs to TXT. Other issues included cryptic reports, lack of visualization tools and lack of published performance measurements. In our testing, we found most annotation reports had a significantly high rate of false positives with some ontologies, such as UMLS and MeSH, used in beta testing (4,5,9). These false positive rates were determined by the curator who did the indexing. Two-letter synonyms, such as “IN” for CD44—Indian Blood Group, also caused high false positives as the OBA concept recognition tool interpreted all incidences of the word ‘in’ as a gene mention for CD44. This issue will be controlled to some degree with the OBA stop words feature, but it could be handled more efficiently. We continue to contribute feedback to OBA developers to enhance the utility of the OBA resource for MGI and for MODs in general.
In May 2009, OBA was released on the NCBO BioPortal, version 2.1, with a new GUI front end and significantly improved annotation statistics and reports, including annotation tag clouds that provide clear visual cues to identify important terms identified in the text (see Figure 3). The web service for this release can process only 100 words at a time. The standalone client version, however, can annotate larger blocks of text and can process 2000 words in <5 min. To optimize curation results, articles should be split into multiple sections (title, abstract, keywords, introduction, methods and materials, results and discussion) and each section numerically ranked to reflect importance. Annotation scores for each gene mention in the article as a whole can be calculated manually by multiplying the relevance score by section weight and summing the scores. We are still exploring how best to take advantage of OBA at MGI. We believe it has great potential as an online curatorial resource, but additional refinements to the user interface, coupled with more flexible data input and reporting options, would make OBA an even more powerful tool for curation workflows.
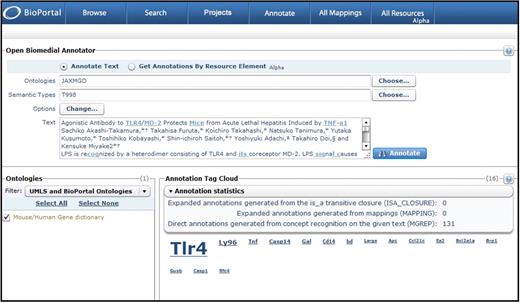
OBA gene mention annotation statistics. In BioPortal Release 2.1, the NCBO Open Biomedical Annotator web service provides visually appealing annotation tag clouds and annotation statistics for plain text submissions processed using the MGI mouse and human gene dictionary (JAXMGD) and a semantic type developed for this dictionary (T998). This screen shot shows the annotation results for the title and abstract of a Journal of Immunology article. The annotation tag cloud correctly identifies Tlr4 and Ly96 as the most prominent genes with the highest relevance scores (scores appear when you mouse over the gene name) discussed in the article. Relevance scores are calculated based on the sum of weights given to each annotation based on the annotation context.
Fraunhofer SCAI ProMiner
ProMiner is a dictionary- and rule-based system from Fraunhofer SCAI that applies sophisticated algorithms for recognizing complex, multi-word named entities in abstracts and full text articles (16). Using TXT, XML, or HTML files as input, ProMiner processes articles in batch and provides in-text visualization tools that clearly depict where terms were found in the context of the original article. When combined with ProMiner's detailed summary reports and hypertext links to source dictionary references, these visualization tools are excellent aids for curators, especially those originally trained to perform article-based editorial tasks. ProMiner incorporates highly curated name and acronym dictionaries, derived from Entrez Gene, SwissProt and cell line resources, such as ATTC and ECACC. It includes built-in algorithms for approximate string matching and ambiguity filters (6,16).
ProMiner differed from other products we evaluated in that it is not a publically available, open-access web service. Instead, it is delivered as a collection of scripts and configuration files that are installed on a server and run from the UNIX or Linux command line. Depending on the technical savvy of the end user, it may require a significant investment of time and training to go live. Therefore, to optimize our test pilot phase with SCAI, we provided ProMiner developers with subset of articles from our gene indexing corpus in TXT, HTML and PDF Format. They, in turn, queried us about our gene indexing preferences to determine how to define ProMiner system parameters to meet our requirements. This enabled the ProMiner development team to more efficiently produce sample tagged documents and reports, based on articles in our pilot corpus, that showcased the system's gene tagging capabilities (see Figure 4) They also quickly responded to our request for a PDF version of the ProMiner. By the end of our pilot project, SCAI had completed an alpha version of ProMiner that could process all PDF files in our test corpus. We are currently beta testing ProMiner for PDFs, and we have asked SCAI to evaluate the MGI mouse dictionary and consider incorporating it into a future release of ProMiner.

Visualization of ProMiner HTML and PDF tagging. The ProMiner tagged-entity visualization feature uses color-coded highlights to identify the source dictionary (mouse or human) of a tagged biological entity. We annotated this Journal of Immunology article with both the PDF and HTML versions of ProMiner to compare tagging styles and performance, then did a manual search for mouse, (highlighted in light blue) in Adobe Acrobat Reader and the HTML browser. As an example of a false positive hit, SLC is tagged as a synonym for the human gene CCL21; the actual reference is to Japan SLC, a mouse strain resources database. In MGI, this article was indexed to mouse genes Tlr4 and Ly96. (A) ProMiner 7.1 for PDF uses layers in Adobe Acrobat to flag gene names in the context of the original article layout. This makes it easier for MGI curators to scan specific sections of articles, such as Materials and Methods section and figure legends, for gene mentions. Due to issues related to PDF text extraction and conversion, this version of ProMiner has difficulty identifying some hyphenated terms and Greek symbols (such as the α in TNF-α), which are correctly tagged in the more mature HTML version of ProMiner. We provide feedback to SCAI on false negatives and false positives in specific documents, so they can enhance ProMiner processing rules and PDF labeling. (B) ProMiner 6.4 for HTML tags gene mentions using hypertext links and numerical references. Here, tan hypertext links indicate gene names matched to human dictionary terms, blue hypertext links are matched to mouse dictionary terms, those with two color tags are found in both dictionaries. Underlying hypertext links point to an object view window that displays the term reference ID and lists all gene synonyms. (C) In ProMiner 7.1 for PDF, human gene dictionary matches are labeled in red, mouse terms are in green, terms found in both dictionaries are highlighted in yellow or orange (there is no meaning associated with these different shades of highlighting; this is a labeling issue that will be addressed in a future update). Link-outs, outlined in red, indicate a popup window containing detailed information about the gene entity, synonyms, and source dictionary, is available by clicking on the term. A link-out for the tagged gene entity TLR4, identified in human and mouse source dictionaries from Entrez Gene, SwissProt and MGI, is shown.
Of all the systems we evaluated, ProMiner most closely fit our gene indexing requirements and met our success criteria. It provided both visualization tools and batch processing capabilities, and it achieved a strong BioCreative F-score of 0.8. Our gene indexers found the list of tagged gene names appended to HTML files and hyperlinked to a table with more information (the gene identifier, source dictionary and synonyms) a particularly helpful reference tool. Although ProMiner is not free, the pricing for academic and non-profit site licenses seemed quite reasonable for a product of this caliber.
The biggest limitation of this solution is that it is not particularly ‘curator-friendly’. However, once the system is installed, the parameters correctly set up, and the primary users trained, running batch processes to annotate articles is not difficult. If we integrate ProMiner into the MGI production system, we will most likely add a graphical user interface to enable curators to access it through our standard editorial interface. In our initial tests, we have annotated sets of 75 full-text articles (averaging 11.5 pages each) in <20 min. (The actual processing time is dependent on the server environment, not the software.) Using ProMiner, curators who gene indexed an average of 50 articles per week can now process 60–70. We expect to further increase productivity gains by refining the annotation project parameters to include a search for the terms mouse, mice, and murine.
Next steps: the future of text mining at MGI
The MGI text mining software evaluation project was an extremely useful, educational exercise for our curation and system administration staff. It gave staff curators the opportunity to see the many different types of text mining systems available and to consider how they might realistically incorporate these tools into their daily workflows. It also provided the MGI GO curation team with a suite of tools they could start using immediately to help streamline gene indexing and GO annotation functions.
Based on the results of this project, we recommended the AIIA gene mention tagger as a general, open-access resource for MGI curators. The MGI GO curation team is continuing to test OBA as tool for screening and prioritizing articles for curation. We have begun formally collaborating with SCAI to evaluate and test ProMiner at MGI during an extended six-month pilot project. We currently have a dedicated server set up to run ProMiner for both HTML and PDF tagging, and have trained three people on staff to run scripts for gene indexing of articles in our annotation pipeline. As part of this collaborative effort, we receive personalized user support from SCAI ProMiner developers and provide detailed feedback to SCAI that they can use to enhance future product releases. At the end of this extended pilot project we will evaluate performance scores using MGI metrics and an updated corpus, and review our collaborative partnership with SCAI. At that point, we will determine whether we want to formally integrate ProMiner into our biocuration workflow and implement it within other curation groups at MGI.
Biomedical text mining continues to be a fast moving field and MGI plans to evaluate new systems as they become available. For example, we are currently testing Reflect, a new open-access NER tool from the European Molecular Biology Laboratory (EMBL) that we learned about after our initial evaluation phase was completed (17). Reflect, which can be run from a web service or as an internet browser add-on, scans HTML documents for gene, protein and small molecule names. Each tagged entity is associated with a pop-up window that contain definitions, images and hypertext links to external web resources, such as Entrez Gene, Ensemble and PubChem. OnTheFly, another promising open-access web-based service, further extends the usefulness of the Reflect server by automatically converting document files in PDF, Microsoft Office, and plain text formats into HTML for Reflect processing (18) (see Figure 5). This tool gives users the option to produce a reference summary of all tagged terms identified in each document. Reflect and OnTheFly are not as customizable as ProMiner, nor can they process articles in batch, but the accessibility and ease of use of the OnTheFly web service interface make it an attractive and readily available tool for our curation staff.

Visualization of OnTheFly and Reflect tagging. OnTheFly enables users to load PDF files for tagging by the EMBL Reflect server. The color coding choices make the marked up document easy to read, however the underlying PDF converter has difficulty translating special characters (such as the Greek letter α in the title) and kerned letters, such as the ‘fi’ in purified and ‘fl’ in inflammation. (A) Tagged entities in this Journal of Immunology article are color coded by type of bioentity and hyperlinked to an entity summary table that provides more detailed reference information. (B) Processing an HTML version of the article using Reflect illustrates OnTheFly faithfully reproduces Reflect annotations. (C) A closer look at the same ‘Materials and Methods’ section in PDF format converted by OnTheFly. Note that Reflect and ProMiner (Figure 4) identified the same gene name mentions and tagged the same false positive match for SLC.
Our goal, now and in the future, is to incorporate bioNLP tools into the MGI biocuration workflow such that they improve the overall efficiency of our curators without compromising the quality of our literature curation. Forums such as the Text Mining Workshop at the Biocuration Conference give curators and text mining software developers an invaluable opportunity to discuss how bioNLP can be applied to effectively resolve issues such as our gene indexing bottleneck. As IE and NER solutions such as those described here become even more flexible and capable of addressing the complex and specialized requirements of different model organism databases, text mining will become an even more invaluable component of any biocuration workflow.
Funding
MGI projects are supported by grants from the National Institutes of Health: National Human Genome Research Institute (grants HG000330, HG002273, HG003622); National Institute of Child Health and Human Disease (grant HD033745) and National Cancer Institute (grant CA089713). This work was funded in part by the University of Maine Graduate School of Biomedical Sciences and the National Science Foundation Integrated Graduate Education and Research Traineeship (IGERT) program (grant 0221625). Funding for open access charge: National Human Genome Research Institute (grant HG000330).
Conflict of interest statement. None declared.
Acknowledgements
The authors thank Lynette Hirschman from the MITRE Corporation for her gracious evaluation of the MGI triage and curation workflow and her text mining system recommendations during this project. The Journal of Immunology granted permission to use the article by Akashi-Takamura et al. titled ‘Agonistic Antibody to TLR4/MD-2 Protects Mice from Acute Lethal Hepatitis Induced by TNF-alpha’, in volume 176, pages 4244–4251, year 2006. Portions of this article appear in Figures 3–5, and were used to illustrate the gene tagging capabilities of different text mining solutions. The information content of this article is property of the respective authors and the Journal of Immunology: Copyright 2006 by The American Association of Immunologists, Bethesda, MD, USA.
References
Appendix
Online resources and product websites
AIIAGMT, Institute of Information Science, Academia Sinica. http://bcsp1.iis.sinica.edu.tw:8080/aiiagmt/index.jsp.
BioCreative: Critical Assessment for Information Extraction in Biology. http://biocreative.sourceforge.net/index.html.
GENIA Project: Mining Literature for Knowledge in Molecular Biology. http://www-tsujii.is.s.u-tokyo.ac.jp/GENIA/home/wiki.cgi
Information Hyperlinked Over Proteins (iHOP). http://www.ihop-net.org/.
IntraPDF PDF to Text Converter.http://www.intrapdf.com/convert_pdf_to_text.htm.
Mouse Genome Informatics (MGI).http://www.informatics.jax.org. Interested developers may contact us directly to discuss availability of specific resources, such as the MGI dictionary of mouse genes and human orthologs.
OBA, National Center of Biomedical Ontology. http://www.bioontology.org.
OnTheFly: Automated annotator for doc(x), pdf, txt, ppt(x), and xls(x) files. http://onthefly.embl.de/index.html.
PDFTron PDF Conversion Tools. http://www.pdftron.com.
ProMiner, Faunhofer SCAI Institut Algorithmen und Wissenschaftliches Rechnen. http://www.scai.fraunhofer.de/bio.0.html?&L=1.
Reflect, European Molecular Biology Laboratory (EMBL). http://reflect.embl.de/.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}