





Abstract
Cancer is a somatic disease. The lack of Indian-specific reference germline variation resources limits the ability to identify true cancer-associated somatic variants among Indian cancer patients. We integrate two recent studies, the GenomeAsia 100K and the Genomics for Public Health in India (IndiGen) program, describing genome sequence variations across 598 and 1029 healthy individuals of Indian origin, respectively, along with the unique variants generated from our in-house 173 normal germline samples derived from cancer patients to generate the Tata Memorial Centre-SNP database (TMC-SNPdb) 2.0. To show its utility, GATK/Mutect2-based somatic variant calling was performed on 224 in-house tumor samples to demonstrate a reduction in false-positive somatic variants. In addition to the ethnic-specific variants from GenomeAsia 100K and IndiGenomes databases, 305 132 unique variants generated from 173 in-house normal germline samples derived from cancer patients of Indian origin constitute the Indian specific, TMC-SNPdb 2.0. Of 305 132 unique variants, 11.13% were found in the coding region with missense variants (31.3%) as the most predominant category. Among the non-coding variations, intronic variants (49%) were the highest contributors. The non-synonymous to synonymous SNP ratio was observed to be 1.9, consistent with the previous version of TMC-SNPdb and literature. Using TMC SNPdb 2.0, we analyzed a whole-exome sequence from 224 in-house tumor samples (180 paired and 44 orphans). We show an average depletion of 3.44% variants per paired tumor and significantly higher depletion (P-value < 0.001) for orphan tumors (4.21%), demonstrating the utility of the rare, unique variants found in the ethnic-specific variant datasets in reducing the false-positive somatic mutations. TMC-SNPdb 2.0 is the most exhaustive open-source reference database of germline variants occurring across 1800 Indian individuals to analyze cancer genomes and other genetic disorders. The database and toolkit package is available for download at the following:
Database URL http://www.actrec.gov.in/pi-webpages/AmitDutt/TMCSNPdb2/TMCSNPdb2.html
Introduction
Cancer is a genetic disease. Accumulation of somatic mutations leads to the development of malignancy. The somatic status of point mutations is ascertained by comparing to its absence in a paired normal sample from the same patient in addition to the reference germline variant public database pool for single nucleotide polymorphisms. Thus, somatic cancer genome analysis necessitates large-scale sequencing not only of the patient tumors but also of paired normal samples along with a public database of normal individuals to reduce false-positive germline variants, especially those prevailing at a low frequency in the given population (1). However, the disparity remains in the proportion of the genomes sequenced per number of individuals in a race/ethnicity inhabiting this planet (2).
Population-specific sequencing projects have contributed immensely to the understanding of the extent of variability and population genome architecture in addition to the global sequencing efforts (1, 3–8). The two major studies recently described the genome sequence variation from 598 and 1029 Indians are the GenomeAsia 100K (9) and the Genomics for Public Health in India (IndiGen) program (10), respectively. These studies have generated a huge corpus of variants from Indian individuals with significant genetic and clinical insights about the population at large. However, the utility of these resources in the cancer somatic analysis settings is yet to be evaluated.
We earlier presented the first open-source variant data from 62 normal exome sequencing samples (11). Here, we present our in-house effort to generate the Tata Memorial Centre-SNP database (TMC-SNPdb) 2.0, as an open-source reference database comprising germline variants from in-house normal samples derived from cancer patients of Indian descent along with the publicly available Indian variant data from the GenomeAsia 100K and IndiGen program. Additionally, using TMC-SNPdb 2.0, we also demonstrate the utility of ethnic-specific resources in a somatic analysis of a whole-exome sequence from 224 in-house tumor samples (180 paired and 44 orphans) of Indian origin.
Materials and methods
Sample collection and ethical approval
The whole-exome data generated from 173 normal samples derived from cancer patients were analyzed in this study. The sample set and study protocols were approved by the IRB and Ethics Committee of Tata Memorial Centre (TMC)—ACTREC. All the ‘normal’ tissue samples were histologically verified by an onco-pathologist to be devoid of cancer tissue.
Development of TMC-SNPdb 2.0 database and toolkit
Variants from 173 individual samples were filtered using the standard quality filters for SNP: QualByDepth (QD) >2, FisherStrand (FS) >60, ReadPos-RankSum <−8, phred-quality (QUAL) <30, StrandOddsRatio (SOR) >3, RMSMappingQuality (MQ) <40, MQRankSum <−12.5; and for insertion and deletion (INDEL), QD >2, FS >200, ReadPosRankSum <−20, QUAL <30. The variants passing were further selected and variant call format (VCF) files obtained from all the normal samples (n = 173) were merged using BCFtools. We applied a coverage filter of ≥5 reads for altered alleles to select the variants. Additionally, we also included the variants with coverage ≤5 but recurrent in ≥5% of the total samples. These constituted the pool of high-quality variants from the in-house normal samples, which were serially depleted against the germline databases: gnomAD (v3.1.2) (4), dbSNP (v151, inclusive of TMC-SNPdb [v1.0]) (12), GenomeAsia (release date: 4 December 2019) (9) and IndiGenomes (release date: 8 January 2020) (10), followed by the somatic variant database; COSMIC (V91) (13). The remaining variants (not previously reported in TMC-SNPdb [v1.0]) constituted the base variant set of the germline dataset TMC-SNPdb 2.0. Unique germline variants from the in-house samples (n = 173), GenomeAsia and IndiGenomes database constitute the final database of TMC-SNPdb 2.0. A schematic of the protocol followed for the development of TMC-SNPdb 2.0 has been summarized in the supplementary figure (Supplementary Figure S1). The variant database has been provided for download as compressed (bgzip) standard VCF file format (version 4.2), along with the tabix index (.tbi) for easy retrieval and usage of the database file across other applications.
Additionally, a toolkit consisting of the database creator from a group of normal (‘dbCreator’), a combiner (‘dbCombiner’)—to combine multiple variant databases and to flag variant database annotation (‘dbAnnotator’)—to denote if the variant is present in the database, functionalities have been developed (Supplementary Figure S2). The UI for this toolkit has been developed using R-Shiny package (https://shiny.rstudio.com/). The scripts for parsing the variant data, merging different indexed VCF files and variant annotation using the variant database are developed in python. Indexed storage and retrieval of the variants are performed using the python-based package of the program, tabix (14). The toolkit package source code (scripts), database files and reference manual for steps to create an in-house normal sample pool and usage information of the TMC-SNPdb 2.0 toolkit are made available with the downloadable package (http://www.actrec.gov.in/pi-webpages/AmitDutt/TMCSNPdb2/TMCSNPdb2.html).
Variant calling and somatic variant analysis pipeline
GATK (v 4.1.8) (15) based best practice variant analysis pipeline (16) was used to generate variant calls from the exome samples. In short, primary genome alignment was performed using BWA-mem (17), to the GENCODE reference genome (GRCh38.p. 12) (18). The pre-processing of read alignments was performed by GATK and variant calling by HaplotypeCaller. The hard (quality) filters with default parameters suggested by GATK for SNP (QD >2, FS >60, ReadPosRankSum <−8, QUAL <30, SOR >3, MQ <40, MQRankSum <−12.5) and INDEL (QD >2, FS >200, ReadPosRankSum <−20, QUAL <30) were applied on the variants obtained and variant positions only having a minimum depth of 5× were selected. Additionally, Mutect2 (v4.2.3) was used for generating somatic variant calls from the tumor samples with matched normal and tumor-only mode along with—panel-of-normals parameter. Combining variants obtained from the normal samples, from all the corresponding tumors (n = 173), a panel of normals (PON) using Mutect2 was constructed and used for subtraction of variants obtained from somatic for variant calling. Among the Mutect2 variants, ones flagged with quality filters (base_qual, map_qual, position and strand_bias) were removed from the analysis. Further, only variants falling within the target capture region (as defined by the BED files of individual capture kits) were considered. In the case of paired tumors, variants from the normal sample were subtracted. Finally, variant calls from GATK and Mutect2 were combined and constituted somatic variant calls.
Annotation of variants using germline and somatic databases
The somatic variants were subject to functional annotation using the VEP tool (19) and vcf2maf (https://github.com/mskcc/vcf2maf) package was used for converting files from VCF to MAF format. The variants were annotated using in-house scripts with the germline databases including gnomAD (4), dbSNP (12), GenomeAsia (9), IndiGenomes (10), TMC-SNPdb 2.0, in-house PON and somatic mutation database, COSMIC (13). VEP-annotated variants reported in the ALFA (20) database as low-frequency germline variants were also flagged. BCFtools (21) was used for combining the VCF files and other variant file manipulations. The overlap statistics between the databases and variants from exome analysis were computed using in-house python scripts.
Results
Characteristics of the germline variants in TMC-SNPdb 2.0 across in-house samples
We processed the variants obtained from in-house normal samples (n = 173) to develop a structured ethnic-specific resource for the germline variant subtraction for a somatic analysis of tumors. High-quality variants obtained from the in-house normal samples were depleted against the germline and somatic databases (described in the Materials and Methods section) to obtain the unique set of germline variants, followed by depletion by germline and somatic variants (Figure 1A). In total, 3 691 587 unique variants were obtained from 173 normal samples in the GATK-Haplotype caller-based variant calling and filtration based on quality filters (defined in the Materials and Methods section). Using the criteria of a minimum depth of 5 for the allele position or recurrence of greater than 5% of samples, 3 508 295 variants were obtained. Depleting the variants using the global population databases (dbSNP and gnomAD) resulted in 339 426 variants further depletion by GenomeAsia and IndiGenomes databases resulting in the retention of 331 326 variants. Overall, 90.55% of variants were found to be overlapping with the four population variant databases. As most of the normal samples used in the analysis were tumor-adjacent normal, depletion by the COSMIC database was performed to rule out any known somatic confounders, wherein 0.42% (n = 1386) variants were found to be overlapping. For the remaining 329 976 variants, finally, VEP tool-based annotation revealed 7.53% variants (n = 24 844) reported as low-frequency germline variants reported in ALFA (20). Finally, 305 132 variants were identified to be novel germline variants from the dataset (Figure 1A), which constituted the TMC-SNPdb 2.0.
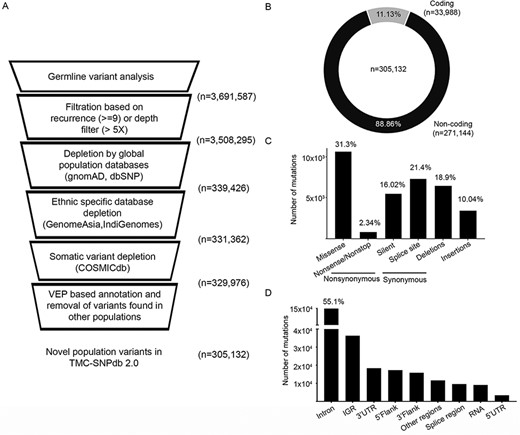
Development of TMC-SNPdb 2.0 and characteristic features of the variants in the database; A) schematic workflow of the steps in the development of the database; the raw variants obtained from the analysis of 173 normal samples were subject to quality/recurrence filter, followed by the depletion of variants found in germline and somatic databases to obtain novel germline variants; B) distribution of coding and non-coding variants; C) distribution of different types of synonymous variants, non-synonymous variants and INDELs; D) proportion of types of non-coding variants, identified in the TMC-SNPdb 2.0. IGR and RNA (in panel D) correspond to the intergenic and non-coding RNA variants in the database, respectively.
The characteristic features of the novel germline variants in TMC-SNPdb 2.0 were coherent with the other variant germline resources. The non-synonymous SNP to synonymous SNP ratio was observed to be 1.95, consistent with the previous version of TMC-SNPdb (11) and literature (3). Of the 305 132 variants, 11.13% (n = 33 988) were found to be within the coding region, whereas 88.86% (n = 271 144) were found to be in the non-coding region of the genome (Figure 1B), consistent with the earlier reports from exome sequencing datasets (22). Among the coding region, missense variants were the dominant type with 31.3% (n = 10 614; Figure 1C). The splice site, insertions and deletion variants were comparatively found to be in the higher proportion, as compared to the previous version of TMC-SNPdb (11), mainly because of the relatively less depletion (as compared to silent variants) by the other population databases (data not shown). Among the non-coding variations, intronic variants were the highest contributors with 49% (n = 149 535) of non-coding variations, followed by intergenic region (IGR) (n = 36 337), 3ʹ UTR (n = 18 374) and others (Figure 1D). Overall, TMC-SNPdb 2.0 catalogs 305 132 novel variants predominant in the Indian population, wherein the majority of the variants (>90%) were with a minor allele frequency of < 5, consistent with the earlier version of TMC-SNPdb (11).
Although the database was created by depleting against the known somatic variants from the COSMIC database, since the normal samples analyzed in the study were from the cancer patients, the presence of possible cancer-predisposing/associated variants within the database could not be ruled out. We performed the deleteriousness prediction on the novel variants to check the proportion of such putative variants. Using SIFT (23) and Polyphen (24) on the 305 132 variants, 4635 (0.01%) and 1919 (0.0006%) variants were assigned the status of deleterious or possibly damaging, by the respective algorithms, suggesting a very small proportion of variants were potentially damaging out of the 305 132 database variants.
Utility of ethnic-specific population germline variant dataset in somatic analysis
A typical cancer-specific somatic analysis utilizes global population databases, such as dbSNP and gnomAD, to flag the possible false-positive somatic status of the variants (25). In addition, to further assess the utility of the ethnic-specific germline variant databases such as TMC-SNPdb 2.0, IndiGenomes and GenomeAsia, in the somatic mutation analysis, we performed variant analysis on in-house 180 paired and 44 orphan tumor samples. Overall median coverage of the tumor samples was observed to be 64.77X. Since whole-exome sequencing was performed using different capture kits, variants satisfying the quality filters (refer to Materials and Methods section) and within the target region of respective kits were selected for further analysis. In total, 1 223 760 unique raw somatic mutations (507 974 from paired and 715 786 from orphan tumors) were obtained from the somatic mutation analysis of 224 tumors analyzed (median mutations = 2881). Raw mutation burden across paired tumor samples was expectedly found to be significantly lower than the orphan tumors (Figure 2A). On the raw somatic variants obtained, we performed a stepwise depletion of the variants using the population germline variant databases (gnomAD, dbSNP), followed by the variant databases representing ethnic-specific Indian/Asian populations (GenomeAsia, IndiGenomes). Variants pooled from all the normal samples, termed as ‘panel of normals’ (PON), were also included in the ethnic-specific variant set. The use of PON is a part of the best practices for the somatic calling workflow, wherein additional low-frequency germline variants in the cohort are removed (26). Major proportion of variants across tumor samples were depleted due to the gnomAD/dbSNP databases (median = 82.61%, range = 21.5–98.4%), with orphan tumors showing relatively higher depletion (median = 96.3%) as compared to depletion in paired tumors (median = 79.5%). Further depletion using GenomeAsia/IndiGenomes/in-house PON, an additional average depletion of 3.44% (range = 0–31.5%, median 2.5%) variants was observed in the tumor samples. In the absence of variants from paired normal tissue, orphan tumors showed significantly higher average depletion of 4.21% (range = 0.27–11.66%, median = 3.23%) as compared to the paired tumors (P-value = 0.001), due to the ethnic-specific variation dataset (Figure 1B). Overall, the variant depletion due to the gnomAD and dbSNP was observed in the range of 64.4–89.8% for paired tumors and 71.9–98.1% for orphan tumors. Depletion using the ethnic-specific variant dataset (GenomeAsia, IndiGenomes, TMC-SNPdb 2.0, in-house PON) showed an additional median reduction of 1.23–5.42% per tumor sample, for paired tumors and 1.9–10.88% per tumor sample, for the orphan tumors. For paired tumors, this constituted a reduction of 2579 false-positive somatic mutations from the exomes analyzed (Table 1) due to the depletion by these ethnic-specific variant datasets.
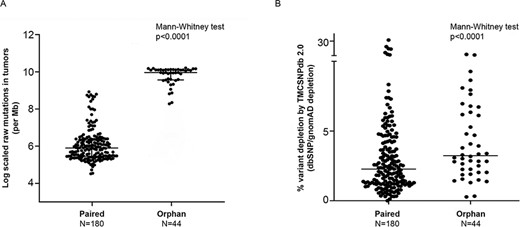
Somatic variant comparison across paired and orphan tumors; A) log-scaled raw mutation count across the paired and orphan exome sequence samples used in the study, B) percent variant depletion by the ethnic-specific germline variant set (GenomeAsia, IndiGenomes, TMC-SNPdb 2.0 and in-house PON created using 173 normal exome samples), over and above gnomAD/dbSNP depletion. Comparison between two groups performed using the Mann–Whitney test.
Statistics of variants obtained from analysis of exome sequencing samples from paired and orphan tumors following depletion of germline variants with the global population variation databases (gnomAD, dbSNP) and Asian/Indian (GenomeAsia, IndiGenomes, TMC-SNPdb 2.0) population germline variant databases, along with variants from the PON derived from 173 in-house normal exome samples
| Unique variants retained upon depletion with databases | |||||
|---|---|---|---|---|---|
| Unique variants | gnomAD + dbSNP | Indian/Asian ethnic-specific databases | Per tumor median % reduction by Indian/Asian databases post dbSNP + GnomAD depletion | Total variants depleted by Indian/Asian databases post dbSNP + GnomAD depletion | |
| Paired Tumor-Normal Samples (n = 180) | 360 352 | 119 608 | 117 029 | 2.27 | 2579 |
| Orphan samples (n = 44) | 378 995 | 88 089 | 86 729 | 3.18 | 1360 |
TMC-SNPdb 2.0 toolkit—a utility package for database access and manipulation
To complement the accessibility of the TMC-SNPdb 2.0 and ease of its incorporation in the somatic analysis pipeline for cancer genomes and exomes, we further developed TMC-SNPdb 2.0 toolkit. The GUI-based toolkit (Supplementary Figure S2) enables researchers to perform the downstream annotation (‘dbAnnotator’) of the somatic variants using the TMC-SNPdb 2.0 or a combination of ethnic-specific Indian/Asian germline variant resources (TMC-SNPdb 2.0, IndiGenomes, GenomeAsia). The database is stored in a tabix-based VCF format, for fast accessibility. The TMC-SNPdb 2.0 toolkit also allows researchers to merge variant database from their in-house normal samples with TMC-SNPdb 2.0 database. This utility is termed ‘dbCombiner’, which also has an inbuilt quality control function that checks for the basic variant quality; using base quality, mapping quality, depth and recurrence criteria, before merging the variants into the database. The merged database can then be accessed using the ‘dbAnnotator’ module. Similarly, the toolkit has an added functionality allowing researchers to create a population database, combining the variant files (VCFs) from the normal samples to create a unique set of variants (‘dbCreator’). The pre-processing steps required to create a combined normal VCF file, installation instructions of the toolkit and the usage description are provided along with the package as a reference manual. The toolkit works cross-platform in Windows and Linux OS. It has been tested on Windows 10/11 and Linux operating systems (Fedora and Ubuntu). Creating a variant database for 100 samples (10 million variants) takes 45 minutes of run-time.
Discussion
We present an updated version of the Indian germline variant database, TMC-SNPdb 2.0, consisting of 305 132 novel Indian population variants from 173 normal whole-exome sequence samples derived from cancer patients, integrated with IndiGenomes and GenomeAsia variant dataset. The high-quality variants—either having a depth of 5 for the alternate allele or present across 5% of the samples, were derived from the 173 normal samples (n = 3 508 295) and overlapped with the global, Asian and Indian germline variant databases, wherein >90% were found to be overlapping pre-reported in these databases. Functional significance of the variants was also evaluated using the prediction algorithms, wherein a small proportion of variants were found to be potentially damaging (0.01% by SIFT and 0.0006% by Polyphen).
Furthermore, using ethnic-specific variant datasets (TMC-SNPdb 2.0, IndiGenomes, GenomeAsia and in-house PON variants), we demonstrate their utility in the somatic mutation analysis pipeline, by analyzing 224 tumor samples (180 paired and 44). Over and above the gnomAD/dbSNP based depletion of depletion (which shows median depletion of 82.6–96.3% variants), an average depletion of 3.4% variants (median = 2.5%) across tumor samples, with orphan tumors showing relatively higher mean reduction of variants by 4.2% (median = 3.23%) per individual tumor was observed. This significant reduction of false-positive somatic mutations due to the low allele frequency ethnic-specific variants demonstrates the utility of using indigenous variant databases in a tumor mutation analysis. Moreover, TMC-SNPdb 2.0 is packaged along with a GUI-based, biologist-friendly toolkit, to allow researchers to integrate the database into their somatic variant analysis pipeline. The TMC-SNPdb 2.0 toolkit can be used to convert variants from an in-house pool of normal samples into a structured variant database, merging user-defined multiple germline variant databases or with custom in-house databases with TMC-SNPdb 2.0. Germline database subtraction is an additional feature of the toolkit that allows annotating the somatic variants (VCF) by TMC-SNPdb 2.0, as well as additional germline databases created by the users.
In a typical cancer-specific mutation analysis, several variants are assigned ‘novel’ or ‘somatic’ status in the context of their absence in population databases. Indigenous, low-frequency variants are confounding factors in the somatic mutation analysis as such variants may be falsely curated as cancer-causing. Deep sequencing of the diverse populations has shown that ethnic-specific or low-frequency variants, which are usually ascribed a status of being deleterious, may be falsely classified due to their frequency of occurrence (1). Sequencing of populations proportionately across diverse ethnicities to cover the maximum disparity human genome holds key to the right assessment of variants.
Characteristic features of the novel variants identified in TMC-SNPdb 2.0 were found to be largely consistent with its predecessor (11), marked by two limitations. First, the normal samples used in the study were derived from cancer patients. The unknown germline predisposing variants occurring at low allele frequency are thus likely to be missed. Second, the paired normal samples, where available, were obtained from sites adjacent to a tumor—after being confirmed as histopathological normal by pathologists—are likely to harbor cancer-associated mutations due to field cancerization (27). To minimize this possibility, we perform depletion of the confirmed somatic variants from the COSMIC database from the TMC-SNPdb 2.0 variant set; thus, the presence of low-frequency cancer-predisposing germline variant cannot be ruled out. The depletion against TMC-SNPdb 2.0 tolerates the loss of rare false negatives but emphasizes the correction for the exclusion of false-positive variants. In summary, we present an updated version of the variant database, TMC-SNPdb 2.0, which includes 305 132 novel germline Indian-specific variants, not reported globally in any of the human genome variation resources. The database is packaged with a toolkit to allow researchers to easily integrate the database into their somatic analysis pipeline. We strongly recommend a collective use of the ethnic-specific variant databases, including IndiGenomes and GenomeAsia, in the somatic variant analysis protocol. This would be especially applicable in analyzing the Indian cancer whole exome and genome and other genetic disorders with a particular focus on the gene–phenotype relationship.
Supplementary data
Supplementary data are available at Database Online.
Acknowledgements
We acknowledge the help received from Dr Sudeep Gupta, Dr Kumar Prabhash, Dr Sudhir Nair, Dr Supriya Chopra and the TMH bio-repository for obtaining the clinical samples.
Funding
This work was supported by an extramural grant from DBT-Virtual National Cancer Institute (VNCI) [BT/MED/30/VNCI-Hr-BRCA/2015] to A.D. The funders had no role in study design, data collection and analysis, decision to publish and preparation of the manuscript.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Data availability
The TMC-SNPdb 2.0 database and toolkit are available at http://www.actrec.gov.in/pi-webpages/AmitDutt/TMCSNPdb2/TMCSNPdb2.html. The database is also available for download via the dbSNP build B156 (https://www.ncbi.nlm.nih.gov/SNP/snp_viewTable.cgi?handle=TMC_SNPDB2) for public access.
References
Author notes
contributed equally to this work.

{kind=link}
{kind=link}