





Abstract
Chemical probes are important tools to investigate the function of proteins, evaluate their potential as therapeutic targets and provide chemical starting points for drug discovery. As a result, a growing federation of scientists aims to generate chemical probes for all human druggable proteins. A diverse array of data typically guides target selection and chemical probe discovery: information on protein function can help prioritize targets, domain architecture can provide insight on druggability, structural data enables molecular design and existing chemical ligands can serve as foundation or inspiration for chemical probe development. But these heterogenous data types are dispersed across a variety of public repositories that are difficult to cross-reference by non-experts. We developed ChemBioPort, an online resource that allows users to combine queries related to the ontology, domain architecture or name of human proteins to produce downloadable tables that integrate information on function, disease association, essentiality, tissue enrichment, domain architecture, structure and chemical ligands of proteins. Users can convert these tables into dendrograms reflecting sequence similarity, onto which they can graphically project all data types, linked via a mouse-click to their original repositories or published articles. This interface will support the growing community of chemical biologists, chemists, cell and structural biologists on their perilous journey from genes to medicines.
Database URL: https://chembioport.thesgc.org
Introduction
The exploration and validation of therapeutic targets is a long and hazardous voyage that often starts with genetic studies linking a gene to a disease and ends with the observed efficacy of a drug in the clinic. Chemical probes—drug-like ligands that potently and selectively engage a target protein in cells—are an important type of tools to guide scientists in this journey, as they can be used to link a protein to a phenotype (1–3). This realization catalyzed pilot initiatives to generate chemical probes for entire protein families such as kinases (4), bromodomains (5), methyltransferases (6) or SLC carriers (7), leading to Target 2035, an ambitious mission where an international federation of scientists aims to produce a chemical probe for all druggable human proteins by the year 2035 (8, 9).
To support the prioritization of targets and the development of chemical probes, we previously created Chromohub and Ubihub, online interfaces integrating data on the disease association, structure and chemical inhibition of proteins involved in chromatin- and ubiquitin-mediated signaling, respectively (10, 11). Because Target 2035 expands beyond these specific areas of human biology, here we present ChemBioPort, a web portal on the structure, function and chemical ligands of all human proteins. ChemBioPort not only expands dramatically the scope of our previous interfaces, but also integrates novel data types, such as gene essentiality from the gnomAD database (12), mass spectrometry-derived tissue enrichment of proteins from the GTEX Consortium (13) or chemical inhibitors from Novartis’ chemogenetic library (14) or the Chemical Probes portal. Novel features are implemented such as ontology- or domain-based search functions, and a mechanism to project heterogenous data types on circular dendrograms derived from multiple sequence alignments of any ensemble of proteins.
ChemBioPort is an intuitive portal to the human proteome that allows medicinal chemists, structural or cell biologists to navigate and integrate diverse arrays of datatypes relevant to chemical probe development such as disease association, tissue expression profiles, gene function and ontology, protein domain architecture, structural homologs or chemical inhibitors.
Data collection
Proteins can be queried based on the gene ontology associated to their enzyme classification number extracted from The Gene Ontology (15, 16) or associated to their structural domains extracted from the European Bioinformatics Institute (EBI) (17) ftp://ftp.ebi.ac.uk/pub/databases/interpro/interpro2go. Protein names, Uniprot IDs and domains are extracted from the EBI’s Uniprot and Interpro databases (17, 18). Disease association is derived from MeSH terms in PubMed as previously described (10) and PubMed records for any gene are extracted from NCBI’s gene2PubMed resource ftp://ftp.ncbi.nlm.nih.gov/gene/DATA/gene2pubmed.gz. Protein structure coverage and structures of bound chemical ligands (molecules with six or more carbons and a molecular weight lower than 1200 Da) are obtained from the Protein Data Bank (PDB) (19). To avoid artifactual chemical inhibitors often found in the literature, we made the choice to use two curated data sources: the chemogenetics library composed of over 4000 compounds manually selected via expert crowdsourcing at Novartis (14) and the Chemical Probes portal (www.chemicalprobes.org). Gene essentiality in human is downloaded from the Broad Institute’s gnomAD database (12). As recommended by gnomAD, a gene is considered essential in human if the upper bound of the oe confidence interval is <0.35. If the lower bound is >0.35, we consider the gene non-essential. If upper bound >0.35 and lower bound <0.35, the gene may be essential which we annotate as ‘Maybe’. Gene essentiality in cancer is derived from CRISPR knockout screens at the Broad and Wellcome Sanger Institutes (20, 21). A gene is considered essential in a given cell line if the CRISPR score is lower than −1.0 [Broad] or its corrected log-fold change is lower than −1.0 [Sanger]. Tissue enrichment of proteins derived from mass spectrometry is obtained from the GTEx Consortium (13): a protein is enriched in a given tissue if its tissue specific (TS) score reaches 2.5 (SD from the mean of the population distribution). If the TS score of a protein is >4 in a tissue and is at least 1.5 higher than the protein’s TS scores in any other tissue, this protein is considered tissue specific. Disease association derived from PubMed data and structural and chemical coverage derived from the PDB are automatically updated weekly while other data are manually updated annually.
Web interface
Any combination of queries is made on a simple online user interface to retrieve human proteins matching specific names or Uniport IDs, gene ontologies or protein domains. For instance, searching for the GO term ‘ligase’ and domain name ‘WD40’ retrieves 37 human ubiquitin ligases containing a WD40-repeat (WDR) domain (Figure 1). Heterogenous lists of protein names or uniport IDs can also be pasted in the query window.
![Querying ChemBioPort. A simple menu (top) is used to produce a downloadable list of proteins (bottom) with annotations extracted from a heterogenous array of data sources. Hovering over ‘information’ icons [i] provides details on the data source and interpretation (top-right).](https://oup.silverchair-cdn.com/oup/backfile/Content_public/Journal/database/2022/10.1093_database_baac088/1/m_baac088f1.jpeg?Expires=1780568231&Signature=GRWmIhwigg8kLGTyLjT5fjRwRazErR9fCn0-~vAGJ9tAkTlhvm6Za2B0FaBtge9VhB6zZ8VrTpEmsXTTZLDEeaWiqgtajTCBczMmkEvemAN7OKj2FYkth64zVjUR6Pnpk9v~mNuAyI5oyQZ123vFrm1IVvRm7Q8~UX94EqHxbB1WGgxxPy-OWSldCUC3LtWgniLlZ1WcrbNNqo~haGHnNqjgjwV3HHLh~J7EMNLgSmW8MPx0dfZsJF3PFpfCU4pDwf0sDLIGFF2iwNCzXB1TKTfJx8DWtQnJg0NJKGY-UUYQsnxP~j4AYiaTKohvd0uLyRxhwoFm6TPmcDvZ95j3kw__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Querying ChemBioPort. A simple menu (top) is used to produce a downloadable list of proteins (bottom) with annotations extracted from a heterogenous array of data sources. Hovering over ‘information’ icons [i] provides details on the data source and interpretation (top-right).
The resulting downloadable table provides the function of each protein (as annotated in Uniprot), the number of articles indexed in PubMed, a good indicator to distinguish highly characterized from underexplored proteins, the number of structures in the PDB (users can opt to only include crystal structures, generally more reliable for structure-based drug design), the number of chemical inhibitors and the number of chemical ligands found with the protein in the PDB. Hovering over or clicking on any of these numbers opens a new window providing further information, such as PDB codes mapped onto the domain architecture of the protein or the chemical structure of ligands, all of which are illustrated in the next section. The next series of data relates to disease association, starting with the number of PubMed entries linking the protein to a specific disease area such as cancer or viral infections, the essentiality of the gene in healthy humans and in cancer, as well as the list of healthy tissues where the protein is enriched or specific.
The output table can be converted on the fly into an evolutionary tree via the automated generation of a multiple sequence alignment produced by ICM (Molsoft, San Diego) (22) on our server where the default Uniprot isoform sequence for each protein is used. Alignments are converted to Newick strings with ICM, which are then represented as dendograms onto which data can be projected, as illustrated in the next section. To avoid the rate-limiting step of generating multiple sequence alignments (which can take up to 1 minutes for ensembles of 80 or more proteins), Newick strings are saved and automatically retrieved when the same query is repeated at a later time or a previously saved query ID (automatically generated for each novel query) is entered.
Application for guiding the development of chemical probes targeting E3 ligases associated with cancer and viral infection
Discovering putative therapeutic targets
Once the evolutionary tree of the 37 E3 ligases listed in Figure 1 is generated, users can easily map onto the tree any combination of data extracted from our database. For instance, histograms can be projected next to each protein name to reflect the number of articles linking the protein to specific disease areas (Figure 2). This bird’s eye view clearly shows for instance that, based on the published literature, CDC20 and DCAF1 are among the most associated with cancer and viral infections respectively. Hovering over an icon brings an enlarged histogram where any bar can be clicked to produce the list of articles associating the gene to a specific disease, each with a link to the corresponding PubMed page (Figure 2).
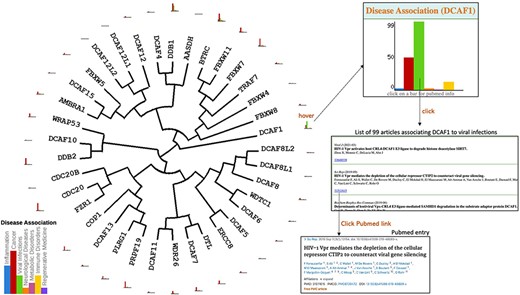
Disease association. Evolutionary dendograms for any table (Figure 1) are automatically generated, onto which data can be projected. Here, histograms representing the number of articles indexed in PubMed linking each gene to specific disease areas are shown. Hovering over histogram icons opens larger bar charts where bars can be clicked to produce the list of articles, each linked to their respective PubMed entry.
The fact that a protein such as CDC20 is associated to cancer based on the literature does not mean that the link is causative. To further investigate this possibility, CRISPR knockout screening data from the Broad and the Wellcome Sanger Institutes (20, 21) reflecting the essentiality of a gene in cancer can be mapped with color-coded icons on the tree (Figure 3). Hovering over an icon next to a gene provides the list of cancer types, the corresponding number of cell lines and essentiality score for the gene of interest for that particular cancer type. Verifying that the data obtained from the two independent institutes is in agreement can add confidence to the result. For instance, CDC20 is one of a handful of WDR-containing E3 ligases that are essential in cancer according to both data sets.
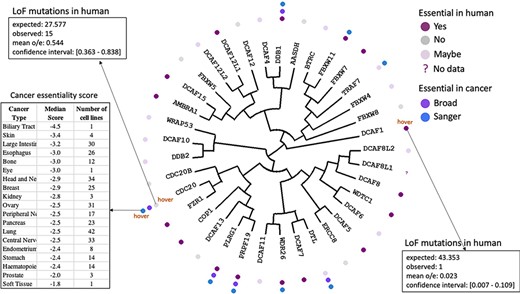
Essentiality in cancer and in healthy individuals. Cancer dependency data derived from CRISPR knockout screens at the Broad and Wellcome Sanger Institutes (20, 21) is mapped next to each protein name in the dendogram. Hovering over icons provides details on essentiality score and number of cell lines representing each cancer type. The essentiality of each gene in healthy individuals derived from observed versus expected LoF mutations (12) is also projected on the tree.
A gene that is essential in cancer may not be a good therapeutic target if it is also essential in healthy tissues. The portal can be used to identify genes essential in the former and not the latter by projecting gene essentiality from the gnomAD database. This data is obtained by comparing the observed and expected numbers of loss-of-function (LoF) mutations in healthy individuals (12): if observed LoF mutations are significantly rarer than expected, it is likely that the gene is essential. This integrative analysis for example reveals that CDC20 (15 observed LoF mutations versus 27 expected) is the only gene in our list that is essential in cancer and not in healthy tissues (Figure 3). On the other hand, DCAF1 is essential (one LoF mutation observed versus 43 expected) and stoichiometric doses of drugs targeting this protein have higher chances of being toxic (Figure 3).
Evaluating chemical tractability
Once a functionally interesting target is identified, a natural next step is to assess its chemical tractability: are structures of the protein revealing a well-defined drug binding site? Are drug-like ligands occupying this site? ChemBioPort rapidly provides answers to these questions. First, the structural coverage of all proteins on the tree can be accessed by hovering over icons next to the protein name. A window appears, listing all structures of the protein in the PDB, and highlighting the structural domains of the protein (defined by their name and their Interpo ID) covered by each PDB code. Hovering over the PDB codes provides the PDB title. Second, structures in complex with drug-like ligands can be mapped onto the tree. Hovering over the corresponding icons displays the chemical structures and clickable PDB codes linked to corresponding pages on the PDB website.
For instance, it becomes immediately clear that a drug-like ligand is found in a structure of CDC20 with PDB code 4N14. Details on the structural coverage indicate that this structure covers the WDR domain of CDC20 (Figure 4). The same tools reveal two drug-like ligands found in the structure of DCAF1 (Figure 4).
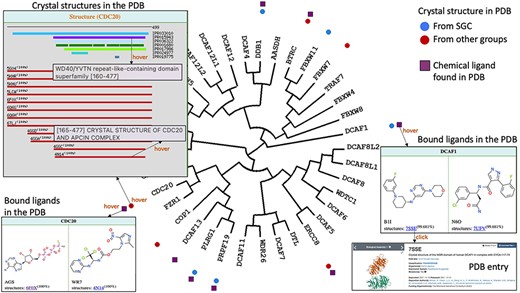
Structural and chemical coverage. Circular icons highlight which proteins have a crystal structure in the PDB. Hovering over an icon provides details on the domain architecture of the protein (including domain name and Interpro ID), the crystallized domains and their clickable PDB codes and titles linked to the associated PDB pages. Data on chemical ligands found in PDB structures were also mapped as square icons. Hovering brings image of the chemical structures and PDB codes.
With just a few clicks, an annotated table of all human WDR-containing E3 ligases was extracted from the database, an evolutionary dendogram was generated, and bird’s eye views of disease association, structural and chemical coverage were produced. Details on the domain architecture and chemical tractability of functionally interesting proteins were rapidly accessed.
A similar integrative analysis can be conducted with any collection of human proteins, selected by name, ontology or structural domain.
Conclusion
The development of a chemical probe requires a multidisciplinary team of scientists that often need to access data outside of their area of expertise. To approach an emerging and underexplored target class, a medicinal chemist may need to intersect disease association with the structural and chemical coverages of the entire protein family; to design a target engagement assay, a cell biologist may want to understand the domain architecture of a protein; to develop a binding assay, a biochemist may need to know whether a peptide was co-crystallized with the protein; to engineer ligand selectivity, a medicinal chemist may need to know the structure of protein homologs. By collecting and integrating functional-, structural- and chemical-genomic data, we believe that ChemBioPort is a unique resource to support the rapidly expending community of scientists engaged in the chemical coverage of the human genome (8, 9).
Acknowledgement
The algorithm used to generate phylogenetic trees from Newick strings was originally written by Xi Ting Zhen.
Funding
The Structural Genomics Consortium is a registered charity (no. 1097737) that receives funds from Bayer AG, Boehringer Ingelheim, Bristol Myers Squibb, Genentech, Genome Canada through Ontario Genomics Institute (OGI-196), EU/EFPIA/OICR/McGill/KTH/Diamond Innovative Medicines Initiative 2 Joint Undertaking (EUbOPEN grant no. 875510), Janssen, Merck KGaA (aka EMD in Canada and USA), Pfizer and Takeda. MS gratefully acknowledges support from NSERC (grant no. RGPIN-2019-04416), CQDM (Quantum Leap-176) and MITACS accelerate (IT13051).
Conflict of interest
None declared.
Data availability
All data is freely accessible at https://chembioport.thesgc.org/.

{kind=link}
{kind=link}
{kind=link}
{kind=link}