





Abstract
The global population surge demands increased food production and nutrient-rich options to combat rising food insecurity. Climate-resilient crops are vital, with millets emerging as superfoods due to nutritional richness and stress tolerance. Given limited genomic information, a comprehensive genetic resource is crucial to advance millet research. Whole-genome sequencing provides an unprecedented opportunity, and molecular genetic methodologies, particularly simple sequence repeats (SSRs), play a pivotal role in DNA fingerprinting, constructing linkage maps, and conducting population genetic studies. SSRs are composed of repetitive DNA sequences where one to six nucleotides are repeated in tandem and distributed throughout the genome. Different millet species exhibit genomic variations attributed to the presence of SSRs. While SSRs have been identified in a few millet species, the existing information only covers some of the sequenced genomes. Moreover, there is an absence of complete gene annotation and visualization features for SSRs. Addressing this disparity and leveraging the de-novo millet genome assembly available from the NCBI, we have developed the Genomic SSR Millets Database (GSMDB; https://bioinfo.icgeb.res.in/gsmdb/). This open-access repository provides a web-based tool offering search functionalities and comprehensive details on 6.747645 million SSRs mined from the genomic sequences of seven millet species. The database, featuring unrestricted public access and JBrowse visualization, is a pioneering resource for the research community dedicated to advancing millet cultivars and related species. GSMDB holds immense potential to support myriad studies, including genetic diversity assessments, genetic mapping, marker-assisted selection, and comparative population investigations aiming to facilitate the millet breeding programs geared toward ensuring global food security.
Database URL:https://bioinfo.icgeb.res.in/gsmdb/
Introduction
Millets, a diverse group of small-seeded grasses with origins primarily in Africa and Asia, have served as essential staple crops in numerous regions globally for centuries [1]. Millets are considered poor people’s food, as they are confined to low-income people [2]. Foxtail millet (Setaria italica), pearl millet (Cenchrus americanus), finger millet (Eleusine coracana), broomcorn millet or proso millet (Panicum miliaceaum), kodo millet (Paspalum scrobiculatum L.), little millet (Panicum sumtrense), barnyard (Indian) millet (Echinochloa frumen), barnyard (Japanese) millet (Echinochloa esculenta), great millet (Sorghum bicolor), adley millet (Coix lacrymajobi), fonio millet (Digitaria exilis) are generally considered as millets.
Millets are renowned for their ability to thrive in harsh growing conditions, including dry and semi-arid climates, making them a valuable food source in areas where other cereal crops may struggle to survive [1, 3, 4]. Among millets, pearl millet has excellent drought and heat tolerance capacity and can be used as a model plant to study drought and heat stress tolerance [5]. It has been known that the root of pearl millet plays an important role in its drought tolerance capacity [6]. Finger millet is also a hardy millet known for its superior drought and salinity stress tolerance [7]. The physiological analysis of 296 broomcorn genotypes on alkali soils (40–80 nM) has proved that Broocorn millet shows high vigor and germination capacity on alkali soils of northern China [8].
Millets are rich in dietary fiber, vitamins (such as niacin, thiamine, and folate), essential amino acids (lysine, methionine, and tryptophan), minerals (including iron, magnesium, and phosphorus), antioxidants [9], and anticancer properties [10, 11]. These nutritional components contribute to the potential health-promoting effects of millets, such as improved digestion, reduced risk of chronic diseases, and enhanced overall well-being [12].
As the global population continues to rise, ensuring food security has become an increasingly pressing concern. However, current staple food crops often fall short of meeting the demands of this growing population. In light of these challenges, millets emerge as a promising solution. Not only are the millets nutritionally rich, but they also exhibit remarkable tolerance to abiotic stresses, making them resilient in varied environmental conditions. This unique combination positions millet as a potential candidates for becoming the new staple crops of the future. To fully unlock the potential of millets, comprehensive genomic information is indispensable. Such data offer insights into the genetic makeup of these crops, shedding light on the underlying mechanisms governing traits crucial for adaptation, yield, and nutritional quality. By delving into the genomic landscape of millets, researchers gain a deeper understanding of genes related to stress tolerance, yield enhancement, and nutritional enrichment, paving the way for more effective plant breeding strategies.
With the availability of the genome sequence of various millet crops, scientists were motivated to analyze the genomic and genetic data for generating large-scale genomic and genic-simple sequence repeats (SSRs) markers for enhanced breeding practices. While strides have been made, a significant void persists in genomic resources for millet species. It is imperative to prioritize filling this gap to unlock the full potential of millets as staple crops. By investing in the expansion of genomic databases and developing novel molecular tools, we can accelerate the pace of millet research and breeding efforts, ushering in a new era of sustainable food production and global food security.
SSRs, commonly referred to as microsatellites or short tandem repeats (STRs), are repetitive nucleotide sequences within DNA, typically composed of one to six base pairs [13]. Based on their repeat type, SSRs are characterized as perfect, imperfect, and compound SSRs [14]. SSRs are widely used as molecular or genetic markers due to their abundant presence, high reproducibility, hypervariability, co-dominant inheritance, and multi-allelic nature [15]. Consequently, SSRs have been employed in various studies, including genetic, evolutionary, molecular breeding, and phylogenetic studies of millet species [16–18]. These markers play a pivotal role in facilitating enhanced breeding practices, enabling targeted selection of desirable traits in millet cultivars. The biological value of SSRs lies in their ability to serve as powerful genetic markers in diverse biological contexts. SSRs consist of STRs of nucleotide motifs scattered throughout the genome. Their inherent polymorphic nature makes them invaluable tools for various genetic analyses, including population genetics, molecular breeding, and evolutionary studies. One of the key advantages of SSRs is their high level of polymorphism, which arises from variations in repeat lengths within populations. This polymorphic nature allows SSRs to reveal extensive genetic diversity within and among populations, making them ideal for assessing population structure, genetic relatedness, and gene flow. Moreover, SSRs are codominant markers, i.e. alleles at a given locus can be distinguished separately in heterozygous individuals. This property enables precise genotype characterization, facilitating the mapping of genes underlying complex traits through linkage and association studies. Additionally, SSRs exhibit multi-allelic variation, providing finer resolution compared to biallelic markers such as SNPs (single nucleotide polymorphisms). This versatility allows SSRs to capture a broader spectrum of genetic variability, enhancing their utility in applications such as marker-assisted selection (MAS), diversity analysis, and germplasm characterization. Furthermore, SSRs are transferable across related species, facilitating comparative genomics and evolutionary studies. Their widespread distribution in the genome and amenability to PCR-based amplification make SSRs accessible and cost-effective markers for researchers across diverse fields of biology.
In summary, the biological value of SSRs lies in their versatility, polymorphic nature, codominant inheritance, multi-allelic variation, and transferability. These characteristics make SSRs indispensable tools for elucidating genetic diversity, understanding evolutionary processes, and facilitating crop improvement and conservation efforts in various biological contexts.
Many bioinformatics databases have been developed for cereal crops like rice [19]. Previously, bioinformatics resources have proven valuable for numerous scientific studies [20–22]. Therefore, multiple databases have also been developed to offer information on SSRs in various millet species. Among millets, foxtail millet is one of the most widely studied millets in terms of genomic research. Five different databases for foxtail millet have been developed, which include databases for transcription factors, microRNAs, genetic markers, and so on [23]. The foxtail millet Marker Database FmMDb [24], which exclusively contains SSR information for S. italica L., the Little Millet Transcriptome Database LMTdb [25], which includes SSRs found in transcriptome sequence of P. sumatrense, and the Millet SSR Database (http://webtom.cabgrid.res.in/millet_ssr_db/team1.php), which encompasses information on genic SSRs in four millet species. The foxtail millet transcription factor database, FmTFDb [23], compasses nearly 2500 transcription factors with their sequence, motif structure, chromosomal localization and expression under different conditions. Pearl millet gene expression and gene function annotation databases have been constructed by a research group in NIPGR, Delhi and Tsugama Lab, Tokyo [26, 27]. The genome sequences of various millets are available, but function and complete annotation still need to be completed or completely absent. The genome sequences are available at chromosomal, scaffold, and contig levels as well.
While existing genomic SSR databases lack representation of various millet species and proper functional annotation of genes, our current research addresses this gap by introducing the Genomic SSRs Millets Database (GSMDB; https://bioinfo.icgeb.res.in/gsmdb/) by encompassing comprehensive information on genic, non-genic, and overlapping genic–non-genic SSRs identified in genomic sequences of seven distinct millet species: foxtail millet (S. italica), pearl millet (C. americanus), finger millet (E. coracana), broomcorn millet or proso millet (P. miliaceaum), great millet (S. bicolor), adley millet (C. lacrymajobi), and fonio millet (D. exilis) (refer to Table 1). Notably, GSMDB offers features such as SSR-specific primer pair design and visualization of SSR locations within their respective genomes, annotated with genes. GSMDB is poised to serve as a valuable tool for applications such as genetic diversity assessment, phylogenetic analysis, species identification, and crop breeding in millets and related species. To date and to the best of our knowledge, no such database has been developed for markers in seven millet crops with detailed gene annotation, and hence, GSMDB fills a significant void as the first comprehensive resource dedicated to structural and comparative genomics of seven millet crops, providing detailed gene annotation.
List of seven millet species used for the analysis
| S. No. | Common name | Scientific name | Assembly accession | Assembly level | Number of chromosome/scaffold/contig | Genome size (Mb) |
|---|---|---|---|---|---|---|
| 1 | Adlay Millet | Coix lacryma-jobi | GCA_009763385.1 | Chromosome | 10 | 1623.704509 |
| 2 | Finger Millet | Eleusine coracana | GCA_021604985.1 | Scaffold | 1196 | 1229.09764 |
| 3 | Fonio millet | Digitaria exilis | GCA_015342445.1 | Contig | 3329 | 757.450839 |
| 4 | Foxtail Millet | Setaria italica | GCA_000263155.2 | Chromosome | 9 | 401.296418 |
| 5 | Great Millet | Sorghum bicolor | GCA_000003195.3 | Chromosome | 10 | 683.645045 |
| 6 | Pearl Millet | Cenchrus americanus | GCA_947561735.1 | Chromosome | 7 | 1778.181882 |
| 7 | Proso Millet | Panicum miliaceum | GCA_002895445.3 | Chromosome | 18 | 837.228188 |
Materials and methods
Data retrieval and processing
Seven genomic sequences of different millet species were retrieved from the National Center for Biotechnology Information (NCBI; https://www.ncbi.nlm.nih.gov/genome) in FASTA file format (Table 1). Notably, feature files (GFF3 or GTF) were unavailable for all the downloaded millet genome sequences. GFF (General Feature Format) files are a type of plain text file used to describe genes and other features of genomic sequences. These files are commonly used in bioinformatics and genomics to store annotations of genomic data, such as gene structures, regulatory elements, and other features. Their importance spans across data integration, research, and the development and use of bioinformatics tools. Consequently, gene prediction of all retrieved genome sequences was conducted with the help of a standalone gene prediction program named AUGUSTUS (https://bioinf.uni-greifswald.de/augustus/) [28] default parameters tailored for plant genomes and annotation of all retrieved genes was done using eggnog-mapper program [29], which is a bioinformatics tool used for functional annotation of proteins based on evolutionary relationship. It contains various inbuilt functional databases including Pfam [30] to predict the functional domains and structural motifs along with their annotation details. The core idea is that proteins with similar evolutionary histories likely have similar functions. The functional descriptions were assigned to all genes in terms of detailed annotation and Pfam.
In Silico SSR mining and primer designing
The MIcroSAtellite Identification tool MISA [31, 32] was used to mine Perfect and compound SSRs in retrieved millet genome sequences using search criteria of minimum length for mono to hexa repeats as 12, and the maximum interruption between two SSRs as 0. These thresholds are chosen to ensure that the identified microsatellites are of sufficient length to be considered biologically significant and to reduce the likelihood of false positives. The output results of MISA for all seven millet species contained information such as repeat number, motif sequence, length of SSR repeat, type of repeats, start and end positions. The distribution of SSRs in genic, non-genic, and overlapping genic–non-genic regions was determined based on the information from genome sequence annotations available in the respective GFF file. The density of SSRs per megabase (Mb) of the genomic sequences of millet species was determined using the following formula:
Density (SSRs/Mb) = Total Number of SSRs/Length of the Genomic Sequence × 1 000 000
The standalone Primer3 tool [33] has been integrated with the database to design primer pairs for all types of SSRs. This integration takes into account essential parameters, including flanking sequence length, primer size, annealing temperature, and product size. Users are granted flexibility within the database to customize these parameters according to their preferences.
Database development
The database housing the identified SSRs was constructed utilizing the open-source relational database management system MariaDB (version 10.4.17). The user interface of the database was created with Bootstrap library, HyperText Markup Language and Cascading Style Sheets to enhance its visual appeal. We used PHP (version 7.3.26), JavaScript, and AJAX in the back-end to facilitate data processing and interaction. Additionally, dynamic tables and visual data representations in the database are generated using JavaScript, DataTable, and Highchart libraries. This online database is an interactive, searchable and downloadable repository for SSRs with unrestricted user access. The flowchart of the study is illustrated in Fig. 1.
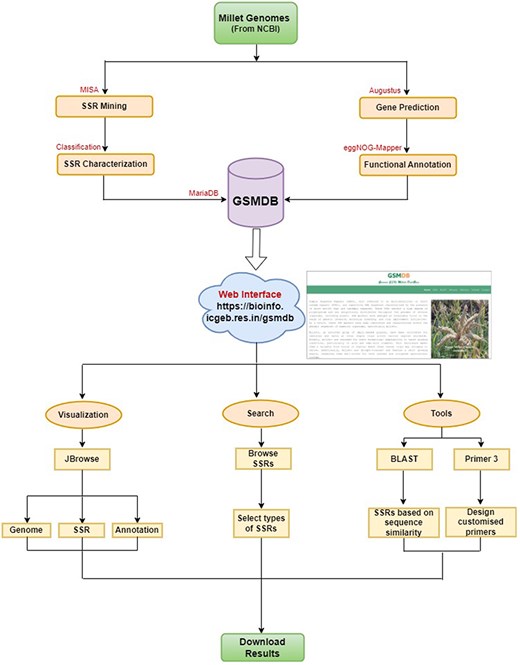
Database architecture and user interface: a complete workflow.
Moreover, the GSMDB incorporates the Basic Local Alignment Search Tool (BLAST) [34] algorithm, which enables a sequence-based search of SSRs within the database. BlasterJS [35] was employed to visualize the BLAST analysis results.
The GSMDB is integrated with JBrowse (version 2.6.3) as the genome browser [36] to show the positions of SSRs and gene annotation along with respective reference sequence of seven millet species. The inclusion of JBrowse features, such as session sharing, importing and exporting sessions, adding custom tracks, plugins, and others, enhances genomic analysis capabilities.
Results
Data statistics
The GSMDB is developed to catalog SSRs identified within the genomic sequences of seven millet species. The GSMDB contains 6 747 645 SSRs, identified with an average density of 7310.6 SSRs per megabase (Mb) in genome sequences of seven millet species (Fig. 2a). Our analysis revealed, in total SSRs, hexanucleotide (4 535 764, 67.22%) repeats as the most abundant, followed by tri- (843 443, 12.5%), tetra- (610 271, 9.04%), di- (273 068, 4.05%), mono- (219 863, 3.26%), and penta- (151 210, 2.24%) repeats. Moreover, compound (51 547, 0.76%) and overlapping compound (62 479, 0.93%) SSRs were also detected in genomic sequences of millets, as depicted in Fig. 2b. Furthermore, our investigation underscores that SSRs are most frequently located within genic (4 674 560, 69.28%) followed by non-genic (2 049 612, 30.38%) regions and overlapping regions that encompass both genic and non-genic domains accounting for 23 473 (0.35%) occurrences (Fig. 2c). Additionally, we have analyzed the top 10 repeat motifs across millet species. It was observed that within the mono-unit, C and G were the most prevalent, while within the di-category, AT and TA were predominant. Similarly, CGC, CGG, and GGC were the most frequently occurring motifs in the tri-category. Only one repeat motif (GGCC) was detected in the tetra-category, whereas no repeat units of penta- and hexa-types were observed among the top 10 motifs. (Fig. 2d).
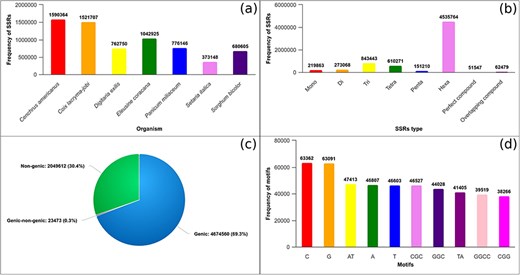
Database statistics: (a) total SSRs identified in genomic sequences of seven millets, (b) total mono-hexa and compound repeats identified in genomic sequences of millets, (c) total SSRs identified in genic, non-genic, and overlapping of genic–non-genic region in millet species, and (d) Top 10 repeat units (motifs) identified across millet species.
Cenchrus americanus displayed the highest frequency of SSRs, with 1 590 364 occurrences with an average density of 894.38 SSR/Mb of the genomic sequence. Hexa- (1 081 887, 68.03%) repeats were most prevalent, followed by tri- (196 639, 12.36%), tetra- (152 559, 9.59%), di- (60 566, 3.81%), penta- (34 879, 2.19%), and mono- (33 865, 2.13%) repeats. Moreover, we also identified compound (11 269, 0.71%) and overlapping compound (18 700, 1.18%) repeats (Table 2A). Distribution of SSR in genomic regions revealed that genic and non-genic regions, with 632 787 (39.79%) and 956 369 (60.14%), respectively. A small number of repeats were also located in the overlapping regions of genic–non-genic (1208, 0.08%) regions of the genome (Table 2B).
Distribution of SSRs in millet species
| A | B | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| S. No. | Species | Mono | Di | Tri | Tetra | Penta | Hexa | Compound | Compound* | Total | Density SSRs/Mb | Genic | Non-genic | Genic-non-genic |
| 1 | Digitaria exilis | 29 551 | 28 004 | 89 060 | 69 119 | 22 288 | 511 635 | 5389 | 7704 | 762 750 | 1007 | 640 726 | 118 483 | 3541 |
| 2 | Eleusine coracana | 77 449 | 55 041 | 122 063 | 77 217 | 22 128 | 672 818 | 8083 | 8126 | 1 042 925 | 848.53 | 851 679 | 185 683 | 5563 |
| 3 | Panicum miliaceum | 25 654 | 31 521 | 106 487 | 77 280 | 19 141 | 503 348 | 5810 | 6905 | 776 146 | 927.04 | 381 864 | 393 003 | 1279 |
| 4 | Setaria italica | 7956 | 11 581 | 46 939 | 33 308 | 8629 | 259 468 | 2459 | 2808 | 373 148 | 929.86 | 324 233 | 47 184 | 1731 |
| 5 | Sorghum bicolor | 13 140 | 33 669 | 72 185 | 73 206 | 15 122 | 461 104 | 6565 | 5614 | 680 605 | 995.55 | 534 852 | 141 650 | 4103 |
| 6 | Cenchrus americanus | 33 865 | 60 566 | 196 639 | 152 559 | 34 879 | 1 081 887 | 11 269 | 18 700 | 1 590 364 | 894.38 | 632 787 | 956 369 | 1208 |
| 7 | Coix lacryma-jobi | 32 248 | 52 686 | 210 070 | 127 582 | 29 023 | 1 045 504 | 11 972 | 12 622 | 1 521 707 | 937.18 | 1 308 419 | 207 240 | 6048 |
| Total | 219 863 | 273 068 | 843 443 | 610 271 | 151 210 | 4 535 764 | 51 547 | 62 479 | 6 747 645 | 922.99 | 4 674 560 | 2 049 612 | 23 473 | |
A: frequency of mono-hexa and compound (overlapping compound*) SSRs; B: frequency of SSRs in genic, non-genic, and overlapping of genic–non-genic regions.
Similarly, SSRs identified in C. lacrymajobi exhibited 1 521 707 SSRs with an average density of 937.18 SSRs/Mb in the genome. Hexa- (1 045 504, 68.71%) exhibited the highest frequency, followed by tri- (210 070, 13.8%), tetra- (127 582, 8.38%), di- (52 686, 3.46%), mono- (32 248, 2.12%), and penta- (29 023, 1.91%) repeats. Compound (11 972, 0.79%) and overlapping compound (12 622, 0.83%) SSRs were also observed (Table 2A). SSRs were identified in genic (1 308 419, 85.98%) regions with higher frequency than non-genic (207 240, 13.62%) and overlapping of genic–non-genic (6048, 0.4%) regions (Table 2B).
Likewise, in E. coracana, 1 042 925 SSRs were identified with an average density of 848.53 SSR/Mb in the genome sequences. Hexa-nucleotides were the most abundant type of SSRs, with 672 818 occurrences, accounting for 64.51% of the total repeats. These were followed by tri- (122 063, 11.7%), tetra- (77 217, 7.4%), di- (55 041, 5.28%), mono- (77 449, 4.43%), and penta- (22 128, 2.12%). Additionally, the presence of compound (8083, 0.78%) and overlapping compound (8126 occurrences, 0.78%) repeats were also observed (Table 2A). In terms of genomic distribution, SSRs in genic (851 679, 81.66%) regions were most frequent, followed by non-genic (185 683, 17.8%) and genic–non-genic (5563, 0.53%) regions of the genomes (Table 2B).
In the analysis of SSRs within the P. miliaceum, 776 146 SSRs were found with an average density of 927.04 SSR/Mb of the genomes. Hexa-nucleotide (503 348, 64.85%) were the most abundant, followed by tri- (106 487, 13.72%), tetra- (77 280, 9.96%), di- (31 521, 4.06%), mono- (25 654, 3.31%), and penta- (19 141, 2.47%). Additionally, both compound (5810, 0.75%) and overlapping compound repeats (6905, 0.89%) were also observed (Table 2A). Concerning their genomic allocation, SSRs were found in both genic (381 864, 49.2%) and non-genic (393 003 occurrences, 50.64%) regions, whereas a small fraction of repeats located in overlapped genic–non-genic (1279, 0.16%) regions in the genome (Table 2B).
Apart from these, 762 750 SSRs were identified in D. exilis genome sequence, and the average density of 1007 SSR/Mb sequence was observed. Hexa-nucleotide repeats (511 635, 67.08%) were found to be the most prevalent, followed by tri- (89 060, 11.68%), tetra- (69 119, 9.06%), mono- (29 551, 3.87%), di- (28 004, 3.67%), and penta- (22 288, 2.92%) repeats. Moreover, repeats of overlapping compounds (7704, 1%) and compound repeats (5389, 0.71%) were also observed (Table 2A). Regarding their genomic distribution, the majority of the SSRs were located in genic regions (640 726, 84%), followed by non-genic regions (118 483, 15.53%), and a small fraction of the repeats were found in overlapping of genic–non-genic (3541, 0.46%) regions in the genome (Table 2B).
In the analysis of S. bicolor, 680 605 SSRs with an average density of 995.55 SSR/Mb were detected. Hexa-nucleotide (461 104, 67.75%) exhibited the highest frequency of the total repeats. Subsequently, tetra- (73 206, 10.8%) were identified, followed by tri- (72 185, 10.61%), di- (33 669, 4.95%), mono- (13 140, 1.93%), and penta- repeats (15 122, 2.22%). Furthermore, both compound (6565, 1%) and overlapping compound repeats (5614, 0.81%) were observed with the least frequency (Table 2A). In the context of their presence in the genomic regions, these SSRs were frequently detected in genic (534 852, 78.58%) regions, followed by non-genic (141 650, 20.81%) and genic–non-genic (4103, 0.6%) regions (Table 2B).
In the genome of S. italica, 373 148 SSRs were mined with 929.85 SSR/Mb density in the genome. Hexa-nucleotide (259 468, 69.53%) were dominating, followed by tri- (46 939, 12.58%), tetra- (33 308, 8.93%), di- (11 581, 3.1%), penta- (8629, 2.31%), and mono- (7956, 2.13%) repeats. Additionally, compound (2459, 0.66%) and overlapping compound repeats (2808, 0.75%) were also identified (Table 2A). Based on their genomic distribution, these SSRs were most prevalent in genic regions (324 233, 86.89%), followed by non-genic regions (47 184, 12.64%). And a minor presence within regions overlapping between genic and non-genic (1731, 0.46%) areas of the genome sequence (Table 2B). So, while the data show a trend where larger numbers of SSRs are observed in species with larger genome sizes, such as C. lacrymajobi and C. americanus, the distribution patterns indicate that SSRs are often concentrated in genic regions across different millet species, regardless of genome size. This suggests that while genome size may influence the total number of SSRs, the specific distribution within genic and non-genic regions remains relatively consistent across species.
Database architecture and utilities
The GSMDB interface comprises seven distinct tabs: Home, SSRs, BLAST, JBrowse, Statistics, Tutorial, and Contact. In the “Home” section, users are introduced to the database with comprehensive details about SSRs and millet species. In the “SSRs” tab, users can delve into SSR data by specifying their preferred millet species. This section is a central hub for accessing and refining queries, allowing users to further specify parameters such as chromosome/scaffold/contig, repeat type, and genomic region after selecting a particular species (Fig. 3a). This enables users to retrieve detailed SSR information in a versatile tabular format. The tabular representation includes essential information such as Motif, Length, Start, End, and Loci details, all aligned with the user’s specific selection criteria. Users also have the option to search for specific motifs within the table using the integrated search bar. Moreover, the interface facilitates data usability by providing options to export table content in various formats through respective buttons. The database also empowers users to design primer pairs targeting the SSRs using the Primer3 tool. Notably, the table includes a “Primer” button that enables users to design primer pairs by selecting the SSR of interest (Fig. 3b). The “Primer” button of the table serves as a pivotal tool, allowing users to customize the flanking sequence length and other fundamental parameters required to design primers for the selected SSR. Once these parameters are specified, users can obtain the results of the designed primer pairs by activating the “Pick primer” button (Fig. 3c). This action triggers the display of comprehensive details regarding the designed primer pairs. Users seeking to employ advanced parameters in their primer design can proceed to the Primer3Plus web page with their selected SSR and flanking sequence by clicking on the link “Click here to proceed to Primer3Plus” (Fig. 3d). The database also provides complete gene annotations of associated SSR upon clicking on respective Loci (Fig. 3e).
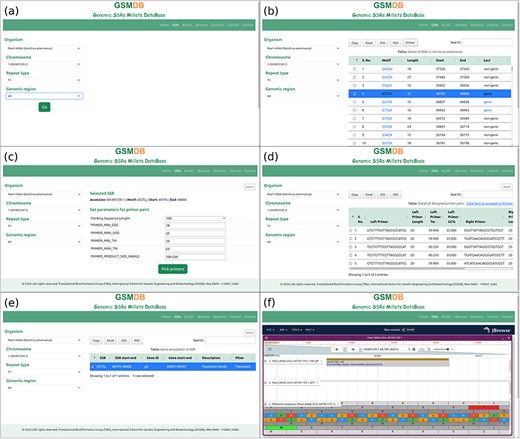
Database snapshots: SSR tab (a) users can select the name of organism and other parameters step by step, (b) detail of SSRs, (c) window to select flanking sequence and parameters to design primer pairs, (d) details of designed primer pairs, (e) gene annotation of respective SSR, (f) JBrowse tab to showcase a comprehensive view, incorporating genome, SSRs, and gene annotations for a more enriched and user-friendly experience.
The “BLAST” tab facilitates a sequence-based search of SSRs within the GSMDB. Users can input nucleotide sequences in FASTA format directly, or alternatively, upload a file (.fasta, .fna, .fa) containing nucleotide sequences for analysis. Upon clicking the “Search” button, the BLAST (blastn) algorithm executes a sequence similarity search against the comprehensive collection of millet sequences cataloged within the GSMDB. The sequence similarity results are displayed in a user-friendly tabular format, outlining matches between the query and genome sequences of millets available in the GSMDB. These matches are based on alignment scores and sequence identities. Users can conveniently review the identified SSRs along with pertinent information such as SSR motif, genomic location, and associated annotations by clicking on the respective “Subject ID” in the table. Additionally, users can visualize sequence similarity results by clicking on the “View Alignment” button. This feature offers a visually appealing, customizable, and highly interactive presentation of aligned sequences along with their corresponding scores, facilitating interactive and intuitive navigation of the alignment results.
The “JBrowse” tab presents SSRs and annotation data on the reference genome for seven millet species. Users can view the Genome, gene annotation, and SSRs tracks simultaneously to comprehensively understand the location of SSRs at desired positions alongside specific gene annotations (Fig. 3f). This tab offers the standard functionality of JBrowse, complete with plugins for more advanced applications. One can easily identify the sequence around upstream and downstream base pairs of selected SSR in the genome browser. It also can be used along with searching “SSRs” tab, which provides the links to respective positions on JBrowse. GSMDB provides interactive zooming and navigation functionalities, allowing users to explore SSR markers at different scales. Users can zoom in to inspect individual SSR loci or zoom out to view broader genomic regions, facilitating a comprehensive analysis of SSR distributions and patterns. Users can view detailed information about individual SSR markers by clicking on them. This feature provides access to key attributes of SSR loci, such as repeat motif, repeat length, genomic coordinates, and associated metadata. It supports search and filtering capabilities, enabling users to quickly locate specific SSR markers or filter markers based on criteria such as repeat motif or genomic region. This feature streamlines data exploration and facilitates targeted analysis of SSR datasets. JBrowse allows users to bookmark specific views or share them with collaborators. This functionality enables seamless collaboration and facilitates the dissemination of SSR marker data across research teams or within the scientific community. Users can also download the desired sequences from JBrowse. Overall, JBrowse provides a comprehensive set of features tailored for SSR databases, empowering users to visualize, analyze, and interpret SSR marker data with precision and efficiency.
The “Statistic” tab offers a graphical representation of the total SSRs accessible within the GSMDB. In contrast, the “Tutorial” page within the database explicates the functionality and interpretation of the available data. Additionally, the database includes a “Contact” tab that offers information about GSMDB developers, and users can convey their inquiries or suggestions to the developers via this tab.
This interactive online database focusing on seven millet species offers a user-friendly experience, enabling users to input queries in the form of genome data and retrieve information based on various search parameters such as chromosome/scaffold/contig, SSR type, motif type, motif length, and more. Users can proceed to design primers for the selected SSRs, and the output is presented in a tabulated format with detailed information, available in csv, txt, or xls formats. Hyperlinks on genic SSRs direct users to gene annotations, enhancing the utility of SSR markers. Each page includes a back button for simplified navigation. The complete statistics of all types of SSRs present in GSMDB are shown in the database. To assist users, a tutorial on navigating the database is provided for a seamless experience. The GSMDB is designed to include more millet genomes to update and integrate the database with future annotations of millet genomes.
Discussion
Developing high-yielding crop varieties essential for addressing global hunger necessitates incorporating molecular markers, which play a pivotal role in breeding strategies, particularly through MAS. Molecular markers are crucial in varietal identification, quantitative trait loci (QTL) mapping, genetic diversity assessment, and comparative genomics. The widespread applicability of markers has led to the generation of extensive DNA marker datasets in various crops. Of the various molecular markers, microsatellites, commonly referred to as SSRs, have gained increased significance in accelerating research. SSRs, found in approximately 10%–20% of genes and promoters in eukaryotic species, exhibit significant variability in length both within and between species. This variability arises from the high frequency of DNA replication errors within SSRs. This variability is important because it contributes to genetic diversity, which is crucial for adaptation and evolution, and influences gene regulation and function, potentially affecting an organism’s phenotype and fitness. The length variability of microsatellites creates a high degree of polymorphism within populations. This genetic diversity is crucial for the survival and adaptability of species, as it provides a pool of variations that can be beneficial in changing environments. Variability in microsatellites can lead to differences in traits that may provide a selective advantage, thereby driving evolutionary processes. Microsatellites located in promoter regions can influence gene expression. Variations in their length can affect the binding efficiency of transcription factors and other regulatory proteins, leading to differences in the level of gene expression among individuals and species. Changes in the length of microsatellites within coding regions can result in alterations in protein structure and function. This can have direct effects on an organism’s phenotype, contributing to variations in traits such as morphology, behavior, and disease susceptibility. The high polymorphism of microsatellites makes them valuable molecular markers for genetic studies. They are used in linkage mapping, population genetics, and biodiversity assessments, providing insights into the genetic structure and evolutionary history of populations. Thus, the length variability of microsatellites within and between species is a fundamental aspect of genetic architecture, influencing everything from molecular mechanisms of gene regulation to large-scale evolutionary patterns. So, this variability makes microsatellites powerful tools for genetic studies because they can distinguish between different genetic lines and track genetic inheritance. Consequently, SSRs, serve as valuable genetic markers due to their polymorphic nature. Despite the emergence of SNPs as a marker resource with advancements in next-generation sequencing technology, SSR markers remain a preferred choice in certain applications. This preference is attributed to the ease of use of SSR markers in molecular laboratories, which require less specialized equipment compared to SNPs. As a result, breeders often opt for SSR markers as their primary choice in marker-assisted breeding. Their importance extends beyond molecular breeding methods, encompassing the realm of diversity assessment in the germplasms of plant species. While numerous SSR marker databases exist for diverse crops, there is a notable gap in comprehensive SSR information for millets despite their nutritional richness and resilience to abiotic stress. Recognizing this gap, GSMDB aims to furnish exhaustive SSR information within the genomic sequences of seven millet species, representing what we believe is the most comprehensive initiative to date in this domain.
The presence of genome sequencing data for millets stands as an invaluable asset, facilitating both comparative and functional genomic studies. Foxtail millet took precedence as the first millet genome to undergo sequencing, primarily due to its modest genome size (∼490 Mb), making it the most suitable candidate for comprehensive whole-genome sequencing and analysis [37]. Historically, until the sequencing of other millet genomes, foxtail millet has been commonly regarded as the reference genome.
The 2012 release of the foxtail millet genome assembly covered approximately 80% of its genome size, encompassing around 38 801 protein-coding genes. Subsequently, in 2017, whole-genome sequencing data for pearl millet and finger millet became available. The initial draft of the pearl millet genome contained an estimated 38 579 genes, with a noteworthy overlap of 15 887 gene families shared with foxtail millet. The distinctive features of the pearl millet genome include a high GC content of 47.9%, often indicative of abiotic stress tolerance [38]. Regarding abiotic stress tolerance, it is important to note that Broomcorn millet exhibits significantly higher water-use efficiency among all cereals. This unique characteristic enables it to produce the highest number of grains using the same amount of water [39]. However, despite the accessibility of genome sequences for millets, there remains a need for comprehensive gene annotation and addressing these annotation challenges is crucial for expediting genomics-based crop improvement programs.
The GSMDB is a valuable repository, providing comprehensive information on genic, non-genic, and overlapping genic–non-genic SSRs within the genomic sequences of millet species with detailed gene annotation. The scrutiny of SSRs in diverse millet species unveiled intriguing patterns concerning repeat type, frequency, and genomic distribution. Hexa-nucleotide repeats were the most abundant across all millet species, comprising a substantial majority (67.22%) of the total SSRs. This prevalence of hexa-nucleotide repeats was also observed with di-nucleotide in maize genomes [40]. The observed variations in the frequency and distribution of identified SSRs among different millet species may be attributed to several factors, including the nucleotide composition of genomic sequences and the specific parameters employed during SSR mining. The dominance of hexa-nucleotide repeats suggests their potential functional significance or selective advantage in the genomes of millet species. Direct functional studies on hexa-nucleotide SSRs are scarce. This could be attributed to several factors. Studying the functional impact of hexa-nucleotide on specific DNA sequences can be complex and requires advanced molecular techniques. SSRs can have diverse functions, including gene regulation, protein structure, and genome stability. Isolating the specific role of hexa-nucleotide SSRs can be challenging. However, the GSMDB provides this opportunity to delve deeply into the roles of specific types of SSRs, which is not possible with other databases developed earlier.
Furthermore, the distribution of SSRs within genomic regions highlighted their preferential location in the genic areas, constituting 69.28% of the total occurrences. This observation is in line with a recent study of maize genomes, also suggesting the distribution of SSRs rich in genic regions [40] in the role of SSRs in the genic areas in influencing gene expression, regulation and potentially contributing to functional diversity. Hexa-nucleotide repeats (64.5%–76.6%) are the most abundant class of SSR in eukaryotes, followed by tri-meric, di-meric, tetra-meric, mono-meric and penta-meric repeats [41]. Also, upon comparative examination of repeat types, it is evident that mono-nucleotide and tri-nucleotide motifs are less prevalent, with penta- and hexa-nucleotide motifs emerging as the most common types across all Oryza species [42]. Studies in other organisms have also shown that the distribution of SSR markers on chromosomes is closely related to gene-rich regions in the plant genomes.
The notable difference in SSR distribution between genic and non-genic regions underscores the importance of understanding the functional implications of SSRs in both coding and non-coding regions of the millet genomes. The presence of SSRs in non-genic regions of plant genomes contributes to various plant traits through several mechanisms Although non-genic SSRs do not directly code for proteins, they can play significant roles in genetic diversity and evolution, gene regulation, genome stability, and adaptation. Non-genic SSRs can influence the expression of nearby genes. They may affect the binding of transcription factors or other regulatory proteins, leading to changes in gene expression levels. For example, SSRs in promoter or enhancer regions can modify the activity of genes, impacting traits such as stress response, growth, and development. Non-genic SSRs can play a role in maintaining genome stability by serving as recombination hotspots. They can facilitate genetic recombination, leading to increased genetic diversity. This increased genetic diversity can enhance a plant’s ability to adapt to changing environmental conditions. Non-genic SSRs are highly polymorphic and can be used as molecular markers in plant breeding programs. They help in the identification of QTL associated with desirable traits. Li et al. (2002) highlighted the role of SSRs in regulatory regions, suggesting that non-coding SSRs can affect transcription, translation, and mRNA stability. The study emphasized that variations in SSR length can impact the expression of nearby genes, influencing phenotypic outcomes. Kelkar et al. (2010) reviewed the impact of SSRs in non-genic regions on disease susceptibility, showing how they can affect gene expression or chromatin structure, influencing the development of various diseases. A study by Gemayel et al. (2010) discussed how SSRs, especially in non-coding regions, could impact epigenetic regulation by altering DNA methylation and chromatin accessibility, indirectly influencing gene expression. So, non-genic SSRs serve as powerful tools for genetic mapping, breeding, and exploring the broader genomic context beyond coding regions.
Cenchrus americanus had the highest SSR frequency among the studied millet species, with 1 590 364 occurrences and an average density of 894.38 SSRs per Mb. This species exhibited a similar pattern of hexa-nucleotide dominance, reinforcing the prevalence of this repeat type. The distribution of SSRs in the genic and non-genic regions of C. americanus revealed a higher frequency in the non-genic areas, suggesting potential roles in regulatory elements or intergenic regions. The variations observed in SSR characteristics among different millet species, such as C. lacrymajobi, E. coracana, P. miliaceum, D. exilis, S. bicolor, and S. italica, emphasize the genomic diversity within the millet family.
Identifying specific SSR patterns in each species provides valuable insights into the evolutionary dynamics and functional aspects of SSRs in individual genomes. The identification of SSR patterns in different species can offer valuable insights into the evolutionary dynamics of SSRs in their genomes. Comparative analysis of SSR types and frequencies across species can reveal patterns of genomic evolution, with variations in SSR distribution and abundance, indicating differences in mutation rates, selection pressures, or genetic drift. Evolutionary conservation of SSRs might suggest important functional roles, while differences in SSR patterns can highlight evolutionary divergence. SSRs’ distribution within genomic regions can provide insights into their roles in genome structure and function. Additionally, SSR variability within and between species can shed light on population genetics and evolutionary dynamics. Phylogenetic analysis of SSR patterns helps in understanding the evolutionary relationships among species. Lastly, SSRs might contribute to adaptive evolution by affecting gene function or regulation in response to environmental pressures. Users can download the desired data from our database.
Conclusion
A meticulously crafted and user-friendly genomic SSRs database, GSMDB, has been established to compile a comprehensive catalog of SSR markers specific to millet species. This database is a valuable reference point for gaining insights into the SSR markers within millet and its closely related species. It contributes significantly to our knowledge of genomic architecture and the functional implications of SSRs in these important crop species. We anticipate that the GSMDB database serves as a central resource for researchers and breeders working on millet improvement, enabling targeted investigations into the roles of SSRs in various genomic contexts and their potential applications in crop breeding and genetic improvement programs In conclusion, we anticipate that this effort will continue to provide pivotal insights, helping mitigate food insecurity and laying the groundwork for a sustainable agricultural system.
Acknowledgements
We acknowledge the grants by the Department of Biotechnology (DBT), Ministry of Science and Technology, Government of India.
Conflict of interest
None declared.
Funding
We acknowledge the grants (BT/IC-06/003/91 and BT/PR40151/BTIS/137/5/2021) by the Department of Biotechnology (DBT), Ministry of Science and Technology, Government of India.
Data Availability
The database website is freely available and accessible at https://bioinfo.icgeb.res.in/gsmdb/
References
Author notes
Both authors contributed equally to this work.

{kind=link}
{kind=link}
{kind=link}