





Abstract
The Saccharomyces Genome Database (SGD; http://www.yeastgenome.org/) organizes and displays molecular and genetic information about the genes and proteins of baker's yeast, Saccharomyces cerevisiae. Mutant phenotype screens have been the starting point for a large proportion of yeast molecular biological studies, and are still used today to elucidate the functions of uncharacterized genes and discover new roles for previously studied genes. To greatly facilitate searching and comparison of mutant phenotypes across genes, we have devised a new controlled-vocabulary system for capturing phenotype information. Each phenotype annotation is represented as an ‘observable’, which is the entity, or process that is observed, and a ‘qualifier’ that describes the change in that entity or process in the mutant (e.g. decreased, increased, or abnormal). Additional information about the mutant, such as strain background, allele name, conditions under which the phenotype is observed, or the identity of relevant chemicals, is captured in separate fields. For each gene, a summary of the mutant phenotype information is displayed on the Locus Summary page, and the complete information is displayed in tabular format on the Phenotype Details Page. All of the information is searchable and may also be downloaded in bulk using SGD's Batch Download Tool or Download Data Files Page. In the future, phenotypes will be integrated with other curated data to allow searching across different types of functional information, such as genetic and physical interaction data and Gene Ontology annotations.
Database URL:http://www.yeastgenome.org/
Introduction
Mutant phenotypes, the outward manifestations of nucleotide sequence changes in the genome, are a key tool in elucidating the functions and roles of gene products. Saccharomyces cerevisiae has a rich store of mutant phenotype information, having been the subject of genetic experimentation for more than half a century in the laboratory and for thousands of years in popular use (1,2; http://biochemie.web.med.uni-muenchen.de/Yeast_Biol/). In recent years, the development of a sophisticated genetic toolkit and the availability of a high-quality genome sequence have facilitated the generation of even more mutant phenotype data from large-scale genetic experiments. Collecting and organizing this range of phenotypic data is among the most important missions of the Saccharomyces Genome Database (SGD; www.yeastgenome.org).
SGD provides comprehensive information about the yeast genome, including the complete genomic sequence as well as the biological roles of chromosomal features and gene products, all curated from the scientific literature (3). The central units of organization of the website are web pages termed the Locus Summary that display information specific to each chromosomal feature (protein- or RNA-coding gene, or other DNA sequence element such as telomere, centromere, etc.). Various tools allow users to search the database in multiple ways in order to find, compare, and analyze sets of chromosomal features. SGD curators and programmers continuously develop new methods for storing, displaying, and searching data in order to keep current with new developments in S. cerevisiae genetic and molecular biology research.
For many years, SGD's curation system for mutant phenotype information consisted of several free-text data fields that could be associated with a reference. The free-text nature of the data made searching and comparing phenotypes a challenge, since the basic concepts were expressed in multiple different ways: for example, the phenotype of heat sensitivity was described using hundreds of disparate phrases, all containing the word ‘heat’, and each linked to only one or a few genes. Furthermore, much of the information was not easily traceable, as it had been derived from unpublished personal communications from researchers who supplied it in the process of reserving new gene names with SGD. To improve the breadth and accessibility of mutant phenotype information in SGD, over the past few years we have developed a system for recording and displaying mutant phenotypes that employs controlled vocabularies for the major concepts while retaining free-text fields to capture experimental details. All of the newly curated information is derived from, and linked to, published references. We describe here the conceptual framework of the curation system, the database and software behind it, its relationship to other phenotype curation systems, and our plans for its future development.
Curation model
What is a mutant phenotype?
The first step in designing a curation system was to define the range of mutant phenotypes that would be curated. For our purposes, a phenotype was defined as the effect of a mutation on any observable or detectable feature of yeast cells, colonies, or cultures. This definition is sufficiently broad to include the most commonly studied yeast phenotypes affecting growth, morphology, and various cellular responses to environmental conditions [see (4) for review]. The aim was to curate these phenotypes as primary observations rather than their interpretations; that is, a growth requirement for adenine would be recorded as auxotrophy, rather than as a defect in adenine biosynthesis, which is the physiological basis for that phenotype.
In determining which observable features to curate, we decided to focus primarily on phenotypes that are detectable at the cellular level (effects on growth, development, and morphology) while also capturing some phenotypes that occur at the molecular level, as long as the observable feature (for example, telomere length) occurs in vivo. We do not capture effects observed only in vitro, such as the decrease in enzymatic activity of a purified mutant protein (this would be curated with a GO annotation). In general, we do not capture ‘self-referential’ phenotypes, meaning the effects of a mutation in a gene on the gene product that it encodes; rather, we capture the consequences of that mutation for other gene products or processes. For example, if a mutation in a gene causes the protein product of that gene to be unstable, we would not curate the instability as a mutant phenotype, but we would curate any effects of the protein instability on the mutant strain, such as a temperature-sensitive growth phenotype.
Another consideration in annotating phenotypes is that each yeast gene product in SGD is annotated with Gene Ontology (GO) (5) terms that describe its molecular function, its subcellular location, and any biological processes in which it is involved. GO annotations may be assigned on the basis of several different types of evidence, including mutant phenotypes, so there can be some overlap between GO annotations and phenotype annotations. The key difference, however, is that while GO terms describe detailed steps in underlying physiological processes, mutant phenotypes represent the overall physiological state resulting from a defect. For some areas of biology, there is congruence between GO annotations for Biological Process and phenotypes: for example, the GO term ‘axial cellular bud site selection’ (GO:0007120) is related to mutant phenotypes affecting budding patterns. In such cases, we would want to annotate all genes reported to be involved in the process, based on mutant phenotype evidence, with both GO and the corresponding phenotype terms. On the other hand, in different areas of biology, there may not be congruence between GO terms and phenotype terms. At the extreme, a gene may have a GO annotation to a process such as ‘DNA replication’, but since this process is essential to life, the related phenotype is ‘inviable’. In other cases, the mutant phenotype is not related in any obvious way to the process in which a gene product is involved: for example, mutations in components of the mitochondrial translation machinery cause the phenotype of mitochondrial genome instability. Annotating mutant phenotypes also allows us to record the fact that a gene may have an extremely indirect effect on a process; in such cases we would not add a GO annotation, since GO annotations should indicate the processes in which a gene product is most directly involved. Thus, while the information conveyed by GO and phenotype annotations may be overlapping and redundant in certain cases, in general the two types of information are complementary. In addition, the phenotype curation system that we have developed allows us to record additional experimental details that are not captured for GO annotations, including information about the underlying mutations (mutant type and allele), the setting in which they occur (strain genetic background and environmental conditions), and the tools used to elicit or assay them (experiment type, chemicals, or reporters).
The curation system described here is currently in use in its complete form for recording mutant phenotypes arising from mutations in single genes. SGD collaborates with the BioGrid database (http://www.thebiogrid.org/) (6) to curate genetic and physical interaction data, and part of the system (a subset of the ‘observable’ terms; see below) is being used in this collaboration for the annotation of phenotypes resulting from genetic interactions between mutations in two genes. The curation system would be easily adaptable for full curation of phenotypes resulting from genetic interactions.
Observables and qualifiers
The core of the phenotype curation system comprises two sets of controlled-vocabulary terms referred to as ‘observables’ and ‘qualifiers’. An observable is a feature of cells, colonies, or cultures, while the qualifier describes the change in that feature relative to wild type. Observables are organized in a hierarchical structure named the Yeast Phenotype Ontology, or YPO. The hierarchical nature of the list facilitates searching, sorting, and comparison of different phenotypes, and in the future will allow the adaptation of existing tools that were originally created to analyze GO annotations, for use with phenotype annotations (see below). The observables, which, like GO terms, have definitions, are words or phrases representing the feature or process that is observed—for example, ‘budding’, ‘filamentous growth’, and ‘resistance to chemicals’. Table 1 shows a subset of the observables, and the entire list may be viewed at http://www.yeastgenome.org/cache/PhenotypeTree.html. Terms are added as necessary, and the YPO is publicly available at the Open Biomedical Ontologies website, http://www.obofoundry.org/.
Observables and qualifiers in the yeast phenotype ontology (YPO)
| Observable |
| • Cellular processes |
| ◦ Chromosome/plasmid maintenance |
| ◦ Intracellular transport |
| ◦ Mitotic cell cycle |
| ◦ Stress resistance |
| • Development |
| ◦ Apoptosis |
| ◦ Budding |
| ◦ Filamentous growth |
| ◦ Lifespan |
| ◦ Necrotic cell death |
| ◦ Sexual cycle |
| ◦ Virulence |
| • Essentiality |
| ◦ Inviable |
| ◦ Viable |
| •Fitness |
| ◦ Competitive fitness |
| ◦ Haploinsufficient |
| ◦ Viability |
| •Metabolism and growth |
| ◦ RNA accumulation |
| ◦ RNA modification |
| ◦ Anaerobic metabolism |
| ◦ Chemical compound accumulation |
| ◦ Chemical compound excretion |
| ◦ Fermentative metabolism |
| ◦ Nutrient utilization |
| ◦ Protein activity |
| ◦ Protein/peptide accumulation |
| ◦ Protein/peptide distribution |
| ◦ Protein/peptide modification |
| ◦ Redox state |
| ◦ Respiratory metabolism |
| ◦ Vegetative growth |
| • Morphology |
| ◦ Cellular morphology |
| ◦ Culture appearance |
| Qualifier |
| • No qualifier |
| • Abnormal |
| ◦ Arrested |
| ◦ Decreased |
| ▪ Absent |
| ◦ Decreased duration |
| Qualifier |
| ◦ Decreased rate |
| ◦ Delayed |
| ◦Increased |
| ◦ Increased duration |
| ◦ Increased rate |
| ◦ Premature |
| • Normal |
| ◦Normal rate |
| Observable |
| • Cellular processes |
| ◦ Chromosome/plasmid maintenance |
| ◦ Intracellular transport |
| ◦ Mitotic cell cycle |
| ◦ Stress resistance |
| • Development |
| ◦ Apoptosis |
| ◦ Budding |
| ◦ Filamentous growth |
| ◦ Lifespan |
| ◦ Necrotic cell death |
| ◦ Sexual cycle |
| ◦ Virulence |
| • Essentiality |
| ◦ Inviable |
| ◦ Viable |
| •Fitness |
| ◦ Competitive fitness |
| ◦ Haploinsufficient |
| ◦ Viability |
| •Metabolism and growth |
| ◦ RNA accumulation |
| ◦ RNA modification |
| ◦ Anaerobic metabolism |
| ◦ Chemical compound accumulation |
| ◦ Chemical compound excretion |
| ◦ Fermentative metabolism |
| ◦ Nutrient utilization |
| ◦ Protein activity |
| ◦ Protein/peptide accumulation |
| ◦ Protein/peptide distribution |
| ◦ Protein/peptide modification |
| ◦ Redox state |
| ◦ Respiratory metabolism |
| ◦ Vegetative growth |
| • Morphology |
| ◦ Cellular morphology |
| ◦ Culture appearance |
| Qualifier |
| • No qualifier |
| • Abnormal |
| ◦ Arrested |
| ◦ Decreased |
| ▪ Absent |
| ◦ Decreased duration |
| Qualifier |
| ◦ Decreased rate |
| ◦ Delayed |
| ◦Increased |
| ◦ Increased duration |
| ◦ Increased rate |
| ◦ Premature |
| • Normal |
| ◦Normal rate |
Only the two highest levels of the observables ontology are shown; most of the terms have more specific child terms. The entire list of observables may be viewed at http://www.yeastgenome.org/cache/PhenotypeTree.html. The entire set of qualifiers is shown. Qualifiers are required with most observables, except those which implicitly include a qualifier (e.g. ‘inviable’).
Observables and qualifiers in the yeast phenotype ontology (YPO)
| Observable |
| • Cellular processes |
| ◦ Chromosome/plasmid maintenance |
| ◦ Intracellular transport |
| ◦ Mitotic cell cycle |
| ◦ Stress resistance |
| • Development |
| ◦ Apoptosis |
| ◦ Budding |
| ◦ Filamentous growth |
| ◦ Lifespan |
| ◦ Necrotic cell death |
| ◦ Sexual cycle |
| ◦ Virulence |
| • Essentiality |
| ◦ Inviable |
| ◦ Viable |
| •Fitness |
| ◦ Competitive fitness |
| ◦ Haploinsufficient |
| ◦ Viability |
| •Metabolism and growth |
| ◦ RNA accumulation |
| ◦ RNA modification |
| ◦ Anaerobic metabolism |
| ◦ Chemical compound accumulation |
| ◦ Chemical compound excretion |
| ◦ Fermentative metabolism |
| ◦ Nutrient utilization |
| ◦ Protein activity |
| ◦ Protein/peptide accumulation |
| ◦ Protein/peptide distribution |
| ◦ Protein/peptide modification |
| ◦ Redox state |
| ◦ Respiratory metabolism |
| ◦ Vegetative growth |
| • Morphology |
| ◦ Cellular morphology |
| ◦ Culture appearance |
| Qualifier |
| • No qualifier |
| • Abnormal |
| ◦ Arrested |
| ◦ Decreased |
| ▪ Absent |
| ◦ Decreased duration |
| Qualifier |
| ◦ Decreased rate |
| ◦ Delayed |
| ◦Increased |
| ◦ Increased duration |
| ◦ Increased rate |
| ◦ Premature |
| • Normal |
| ◦Normal rate |
| Observable |
| • Cellular processes |
| ◦ Chromosome/plasmid maintenance |
| ◦ Intracellular transport |
| ◦ Mitotic cell cycle |
| ◦ Stress resistance |
| • Development |
| ◦ Apoptosis |
| ◦ Budding |
| ◦ Filamentous growth |
| ◦ Lifespan |
| ◦ Necrotic cell death |
| ◦ Sexual cycle |
| ◦ Virulence |
| • Essentiality |
| ◦ Inviable |
| ◦ Viable |
| •Fitness |
| ◦ Competitive fitness |
| ◦ Haploinsufficient |
| ◦ Viability |
| •Metabolism and growth |
| ◦ RNA accumulation |
| ◦ RNA modification |
| ◦ Anaerobic metabolism |
| ◦ Chemical compound accumulation |
| ◦ Chemical compound excretion |
| ◦ Fermentative metabolism |
| ◦ Nutrient utilization |
| ◦ Protein activity |
| ◦ Protein/peptide accumulation |
| ◦ Protein/peptide distribution |
| ◦ Protein/peptide modification |
| ◦ Redox state |
| ◦ Respiratory metabolism |
| ◦ Vegetative growth |
| • Morphology |
| ◦ Cellular morphology |
| ◦ Culture appearance |
| Qualifier |
| • No qualifier |
| • Abnormal |
| ◦ Arrested |
| ◦ Decreased |
| ▪ Absent |
| ◦ Decreased duration |
| Qualifier |
| ◦ Decreased rate |
| ◦ Delayed |
| ◦Increased |
| ◦ Increased duration |
| ◦ Increased rate |
| ◦ Premature |
| • Normal |
| ◦Normal rate |
Only the two highest levels of the observables ontology are shown; most of the terms have more specific child terms. The entire list of observables may be viewed at http://www.yeastgenome.org/cache/PhenotypeTree.html. The entire set of qualifiers is shown. Qualifiers are required with most observables, except those which implicitly include a qualifier (e.g. ‘inviable’).
The qualifiers that accompany observables are used to describe how the observable feature in the mutant differs from wild type: for example, ‘increased’, ‘abnormal’, ‘absent’, and ‘decreased duration’. Qualifiers are also organized in a hierarchical fashion, with the top-level terms ‘abnormal’ and ‘normal’; at present, 13 qualifiers are in use (Table 1). Qualifier terms are incorporated into the YPO, and are distinguished from observables by their namespace, or context, in the ontology. The ‘normal’ qualifier, indicating that the observable feature is the same in the mutant as in wild type, is not used routinely, since it is implicit when describing a mutant phenotype that all other features of the mutant are normal. However, it is used for recording cases where an expected mutant phenotype is not observed, given what is known about the function of a gene. Additionally, the ‘normal’ qualifier is used to record and compare phenotypes resulting from genetic interactions to those arising from single gene mutations, since the severity of a phenotype of a double mutant compared to that of either single mutant can reveal functional interactions between the gene products. In these cases when the phenotype of the double mutant strain is only interpretable within the framework of the single mutant phenotypes, the ‘normal’ qualifier may be used to record the phenotype of the single mutant.
In nearly every case, the core of a phenotype annotation in SGD consists of an observable combined with a qualifier. The few exceptions to this rule (currently, 7 terms out of a total of more than 190) are ‘classical’ phenotypes such as ‘sterile’, ‘petite’, or ‘inviable’, in which the experimental observations are expressed as a single word or phrase. These expressions have a long history in the literature and are obvious search criteria; any phenotype curation system that did not include them would be considered incomplete by yeast researchers. Although the classical phenotypes are not precisely equivalent to the rest of the YPO terms, in that they implicitly include both qualifier and observable and may map to multiple qualifier and observable pairs, they are essential to the user-friendliness of the system for the yeast community.
Experiment type
The ‘experiment type’ namespace of the YPO captures the methods used to generate and analyze the phenotype, as well as the ploidy of the strains involved. The two major experiment types are ‘classical genetics’ and ‘large-scale survey’. Haploidy is implicit, since most genetic experiments are performed using haploid strains. The ‘classical genetics’ experiment type indicates a typical small-scale laboratory experiment in which the phenotypes of one or several haploid mutants are analyzed; the sub-types ‘heterozygous diploid’ and ‘homozygous diploid’ are available where appropriate. The ‘large-scale survey’ experiment type is used for high-throughput screens involving large collections of mutant strains whose design and construction depended on knowledge of the genome sequence. Sub-types of ‘large-scale survey’ indicate whether the experiment employs a systematic mutation set such as the genome-wide deletion mutant collection (7), or involves a technique such as competitive growth, in which a collection of mutant strains is pooled and grown under defined conditions in which they compete with each other or with the wild-type parent for many generations (8,9). The complete list of experiment types is shown in Table 2. Additional fields applicable to large-scale experiments allow the entry of a free-text description of experimental methodology, and of numerical values used as metrics of the phenotype such as fitness score, budding index or growth rate inhibition coefficient.
Experiment types in the YPO
| Phenotype assays |
| • Classical genetics |
| ◦ Heterozygous diploid |
| ◦ Homozygous diploid |
| • Large-scale survey |
| ◦ Competitive growth |
| ▪ Heterozygous diploid, competitive growth |
| ▪ Homozygous diploid, competitive growth |
| ◦ Heterozygous diploid, large-scale survey |
| ◦ Homozygous diploid, large-scale survey |
| ◦ Systematic mutation set |
| ▪ Heterozygous diploid, systematic mutation set |
| ▪ Homozygous diploid, systematic mutation set |
| Phenotype assays |
| • Classical genetics |
| ◦ Heterozygous diploid |
| ◦ Homozygous diploid |
| • Large-scale survey |
| ◦ Competitive growth |
| ▪ Heterozygous diploid, competitive growth |
| ▪ Homozygous diploid, competitive growth |
| ◦ Heterozygous diploid, large-scale survey |
| ◦ Homozygous diploid, large-scale survey |
| ◦ Systematic mutation set |
| ▪ Heterozygous diploid, systematic mutation set |
| ▪ Homozygous diploid, systematic mutation set |
The complete list is shown. Haploidy is implicit unless otherwise specified.
Experiment types in the YPO
| Phenotype assays |
| • Classical genetics |
| ◦ Heterozygous diploid |
| ◦ Homozygous diploid |
| • Large-scale survey |
| ◦ Competitive growth |
| ▪ Heterozygous diploid, competitive growth |
| ▪ Homozygous diploid, competitive growth |
| ◦ Heterozygous diploid, large-scale survey |
| ◦ Homozygous diploid, large-scale survey |
| ◦ Systematic mutation set |
| ▪ Heterozygous diploid, systematic mutation set |
| ▪ Homozygous diploid, systematic mutation set |
| Phenotype assays |
| • Classical genetics |
| ◦ Heterozygous diploid |
| ◦ Homozygous diploid |
| • Large-scale survey |
| ◦ Competitive growth |
| ▪ Heterozygous diploid, competitive growth |
| ▪ Homozygous diploid, competitive growth |
| ◦ Heterozygous diploid, large-scale survey |
| ◦ Homozygous diploid, large-scale survey |
| ◦ Systematic mutation set |
| ▪ Heterozygous diploid, systematic mutation set |
| ▪ Homozygous diploid, systematic mutation set |
The complete list is shown. Haploidy is implicit unless otherwise specified.
Mutant information
The ‘mutant type’ namespace of the YPO captures the impact of the mutation on the activity of the gene product, and not the molecular details of each mutation. Example mutant types are ‘null’, ‘conditional’, ‘overexpression’ or ‘repressible’ (see Table 3 for the complete list, with definitions). The curation system also includes the ability to record allele names (e.g. ‘act1–157’) and details about the alleles (e.g. ‘D157E’, denoting a change at codon 157 from aspartic acid to glutamic acid). While we cannot comprehensively record information about the thousands of mutant alleles that are available, we make an effort to capture representative alleles reported in each study.
Mutant types in the YPO
| Mutant type | Definition |
|---|---|
| Activation | The mutation increases the normal activity of the gene product |
| Conditional | The activity of the gene product is normal under some conditions and altered under others |
| Dominant negative | The mutation results in a gene product that interferes with the function of the normal, wild-type gene product |
| Gain of function | The mutation confers a new activity on the gene product |
| Misexpression | The mutation results in expression of the gene product at a developmental stage, in a cell type, or at a subcellular location different from that at which the wild-type gene is expressed |
| Null | The mutation completely abolishes the function of the gene product |
| Overexpression | The mutation causes expression of the otherwise normal gene product to higher levels than wild type |
| Reduction of function | The mutation reduces the activity of the gene product |
| Repressible | The mutation causes a reduction in levels of the gene product, often through the use of a repressible promoter |
| Unspecified | A curator has tried to determine the mutant type but it is not specified in the article |
| Mutant type | Definition |
|---|---|
| Activation | The mutation increases the normal activity of the gene product |
| Conditional | The activity of the gene product is normal under some conditions and altered under others |
| Dominant negative | The mutation results in a gene product that interferes with the function of the normal, wild-type gene product |
| Gain of function | The mutation confers a new activity on the gene product |
| Misexpression | The mutation results in expression of the gene product at a developmental stage, in a cell type, or at a subcellular location different from that at which the wild-type gene is expressed |
| Null | The mutation completely abolishes the function of the gene product |
| Overexpression | The mutation causes expression of the otherwise normal gene product to higher levels than wild type |
| Reduction of function | The mutation reduces the activity of the gene product |
| Repressible | The mutation causes a reduction in levels of the gene product, often through the use of a repressible promoter |
| Unspecified | A curator has tried to determine the mutant type but it is not specified in the article |
The complete list is shown, with definitions.
Mutant types in the YPO
| Mutant type | Definition |
|---|---|
| Activation | The mutation increases the normal activity of the gene product |
| Conditional | The activity of the gene product is normal under some conditions and altered under others |
| Dominant negative | The mutation results in a gene product that interferes with the function of the normal, wild-type gene product |
| Gain of function | The mutation confers a new activity on the gene product |
| Misexpression | The mutation results in expression of the gene product at a developmental stage, in a cell type, or at a subcellular location different from that at which the wild-type gene is expressed |
| Null | The mutation completely abolishes the function of the gene product |
| Overexpression | The mutation causes expression of the otherwise normal gene product to higher levels than wild type |
| Reduction of function | The mutation reduces the activity of the gene product |
| Repressible | The mutation causes a reduction in levels of the gene product, often through the use of a repressible promoter |
| Unspecified | A curator has tried to determine the mutant type but it is not specified in the article |
| Mutant type | Definition |
|---|---|
| Activation | The mutation increases the normal activity of the gene product |
| Conditional | The activity of the gene product is normal under some conditions and altered under others |
| Dominant negative | The mutation results in a gene product that interferes with the function of the normal, wild-type gene product |
| Gain of function | The mutation confers a new activity on the gene product |
| Misexpression | The mutation results in expression of the gene product at a developmental stage, in a cell type, or at a subcellular location different from that at which the wild-type gene is expressed |
| Null | The mutation completely abolishes the function of the gene product |
| Overexpression | The mutation causes expression of the otherwise normal gene product to higher levels than wild type |
| Reduction of function | The mutation reduces the activity of the gene product |
| Repressible | The mutation causes a reduction in levels of the gene product, often through the use of a repressible promoter |
| Unspecified | A curator has tried to determine the mutant type but it is not specified in the article |
The complete list is shown, with definitions.
Strain background
There are many strains of the species S. cerevisiae, some with long pedigrees from decades of propagation in the laboratory. Although genomic sequencing has revealed that there may be many nucleotide sequence differences between strains (10), the overall physiological differences between them can be relatively subtle, such as differing efficiency in the utilization of particular carbon sources. In curating functional information, such as assigning GO annotations, the strain background used for experimentation is not captured, since it is unlikely that the normal function, role, or subcellular location of a gene product will differ between strains. However, for a subset of genes, strain-to-strain differences between the mutant phenotype of a particular gene can be quite dramatic. For example, the MYO1 gene is essential in the W303 genetic background, but not in the S288C background (11). Because of this possibility, strain background information is collected as part of the phenotype annotation. An initial list of 12 major strain backgrounds is currently in use and will be updated as necessary.
Additional details
The curation system also incorporates the ability to record other types of information that are relevant to the mutant phenotype. ‘Condition’ refers to environmental conditions under which the mutant phenotype is observed, such as growth medium or temperature. Standard conditions for S. cerevisiae consist of rich medium containing 2% glucose as a carbon source (YPD), at 30°C; in general, these standard conditions are implicit and only differences from these conditions are recorded.
‘Chemical’ refers to any chemical relevant to the phenotype, and is most often used to record a drug or chemical stress to which the mutant may display resistance or sensitivity, but may also be used to record alternative carbon sources, required amino acids, or other substances involved in assaying the phenotype. Chemicals are recorded using ChEBI IDs from the Chemical Entities of Biological Interest ontology (ChEBI; http://www.ebi.ac.uk/chebi/). SGD curators participate in the development of ChEBI during the process of curation, routinely requesting new terms for substances used in yeast research that are not currently represented in the ChEBI ontology.
Other entities that are used to assay the mutant phenotype, most often proteins, are recorded as a ‘Reporter’. For example, maturation of carboxypeptidase Y (Prc1p) is commonly used to indicate activity of the vacuolar protein sorting pathway. Finally, ‘Details’ provides a free-text field for information that will help users understand the phenotype or find it in a search. For example, the observable-qualifier pair ‘cell shape: abnormal’ for the SCS2 gene is accompanied by the Detail ‘highly elongated cells’, which provides additional descriptive information and also enables users searching for mutations conferring ‘long’ or ‘elongated’ cell shapes to find such phenotypes.
Curation workflow and quality control
Mutant phenotype curation is routinely performed by all SGD curators (approximately nine full-time equivalents) as part of our normal curation pipeline. We search PubMed weekly, in a procedure that has both automated and manual components, to ensure that we identify all the literature relevant to S. cerevisiae genes and proteins. The most recent literature is our highest priority for curation—in particular, the papers with new characterizations of previously unknown genes.
In addition to keeping up with mutant phenotype curation from these high-priority papers, we have performed targeted efforts to populate SGD's mutant phenotype data since implementing the current curation system. Initially, we gathered all the previously curated phenotype information in SGD, most of which was unreferenced, and set out to find those mutant phenotypes in the published literature and re-curate them using the new system. In addition, drawing on the collective knowledge of SGD staff and advisors we compiled a list of important early mutant studies in which classes of yeast genes were genetically characterized, such as the cell division cycle (CDC) or sterile (STE) mutants, and added the mutant phenotypes from those papers to the database. Currently, we are working towards curating phenotypes from our entire collection of papers that contain large-scale phenotype data: we have about 220 such papers, and have curated the phenotype data sets from half of them. Going forward, our aim is to work towards comprehensive coverage of mutant phenotypes by ensuring that we have recorded representative phenotypes for each gene. There are currently about 1000 genes that lack mutant phenotype annotations from small-scale experiments, but are associated with six or more papers that we have broadly categorized as containing some information about mutations or phenotypes. Annotating mutant phenotypes for this set of genes will be our next goal.
During the 2.5 years since this phenotype curation system was implemented at SGD, we have annotated phenotypes from 1860 papers, covering 94% of protein-coding genes, at an average rate of 59 ‘classical’ and 3 large-scale papers per month. At this rate, we should be able to attain comprehensive phenotype coverage for the S. cerevisiae genome, while keeping up with the new literature, over the next 2–3 years.
Since this curation system is relatively new, our automated quality control at present consists mainly of checks on the data themselves (see below) in order to ensure that they are complete and correctly formatted. In the future, we will devise ways of leveraging other data in SGD to evaluate the completeness of the mutant phenotype data. For example, gene ontology annotations in SGD are thorough and comprehensive, since we have been performing this curation for many years and have given it our highest priority. We will review existing GO annotations made with the ‘Inferred from Mutant Phenotype’ evidence code to determine if related phenotype annotations should be made. We will also review GO annotations to biological process terms that are similar to YPO observables, to check whether mutant phenotype annotations to those observables have been completed.
We have several non-automated mechanisms in place to ensure that phenotype curation is consistent between different curators. We actively maintain extensive help documentation that explains how to record various types of information. We meet weekly to discuss issues that have arisen during curation, and come to a consensus on the best way to deal with them. Finally, several times a year all group members independently curate the same paper, then compare and discuss the results, in order to ensure that each curator is extracting the same breadth and depth of information from each paper and recording it in a consistent manner. The reproducibility of the core curation, i.e., the choice of observables and qualifiers, has been very good. Curation of the additional details is becoming more consistent as SGD curators become more familiar with the curation system. With each curation consistency exercise we have added to our curator help documentation, and we have also added several checks to the curation interface to ensure that all of the required data are entered.
Database and software
Data tables
Two types of information related to phenotype curation are stored in the SGD relational database: the controlled vocabularies, including the phenotype ontology (YPO); and the phenotype annotations that link the controlled-vocabulary terms and additional free-text information to genes and references.
The vocabularies are stored in a set of tables originally designed by CHADO (12) for generic controlled vocabulary data (Figure 1). These tables store the controlled vocabulary terms, the relationships between these terms, and any synonyms for the terms. The observables, qualifiers, experiment types, mutant types and strain backgrounds are stored in these controlled vocabulary tables, as well as the ChEBI ontology terms (which are updated monthly from the ChEBI project).
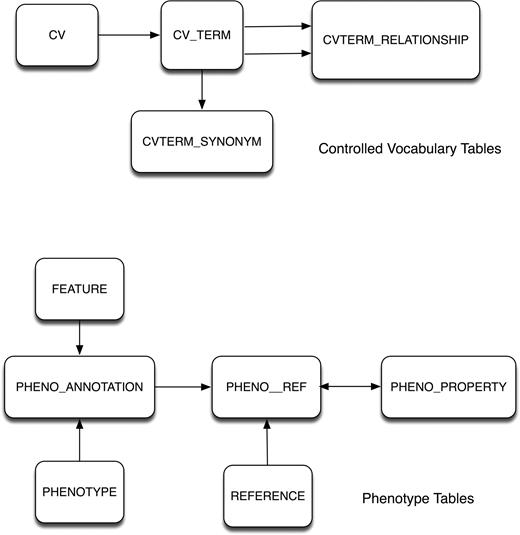
Database schema for mutant phenotype-related data. Top, the controlled-vocabulary tables adapted from the CHADO schema. Bottom, the phenotype data tables.
The phenotype annotation data are stored in a set of tables that link a feature with a phenotype as identified in a reference. The PHENOTYPE table stores the core phenotype description: the observable, qualifier, experiment type and mutant type. A particular combination of these four attributes is linked to a gene or genes in the PHENO_ANNOTATION table. Additional attributes or properties of the phenotype (Allele, Strain background, Condition, Reporter, Details, etc.) are stored in a separate property table that is associated with a specific feature-phenotype combination through one or more references. Figure 1 depicts the general schema; detailed diagrams are available at SGD (http://www.yeastgenome.org/schema/Schema.html).
Before phenotype data are inserted or updated, database triggers validate that the entered data values exist in the controlled vocabulary tables. This allows the actual phenotype values to be stored in the phenotype tables, rather than the conventional relational database practice that would use surrogate foreign keys to physically link to the controlled vocabulary tables. Triggers ensure data integrity and have the added benefit of reducing the complexity of data retrieval by minimizing table joins.
Software
A number of applications were created to assist the curators and the users of SGD with the new phenotype system. A CGI curation interface was developed in Perl to allow curators to easily insert, update, and delete phenotype annotations via the web. In addition, because many phenotypes in the previous free text format had to be converted to a new system, a script was written to load the information onto the database from a flat-text file so that curators could begin work before the curation interface was operational. The SGD checking scripts, which run periodically, were modified to ensure integrity of the phenotype data. Methods were added to automatically create a tab-delimited text file (phenotype_data.tab), containing all currently available phenotype data, that is available for download by SGD users. Finally, to incorporate phenotype information into the Locus Summary pages and into a phenotype-specific search interface, a Model-view-controller (MVC) system was implemented in Perl using Template::Toolkit (http://search.cpan.org/dist/Template-Toolkit/), DBIx::Class (http://search.cpan.org/dist/DBIx-Class/), and CGI::Application (http://search.cpan.org/dist/CGI-Application/). The ‘Model’ part of the system comprises the data from the database, the ‘View’ uses Template::Toolkit for displaying the information, and the ‘Controller’ uses both DBIx::Class and CGI::Application for data retrieval and manipulation.
Curation interface
The phenotype curation interface, accessed by entering a gene or feature name, displays a simple table containing all existing phenotypes curated for that gene, and uses scroll boxes containing the mutant type, experiment type, and qualifier hierarchies for entering new phenotype annotations. The YPO and strain background-controlled vocabularies are accessed via pop-up windows, and there is a link to open the ChEBI website in a new browser window to look up ChEBI IDs for chemicals or other relevant substances. The controlled vocabulary values may be selected by clicking on terms in the scroll boxes or pop-up windows, and text entry boxes allow curators to enter the reference number and free-text information. If one phenotype from one reference is applicable to multiple genes, the curator may enter additional gene names, and the phenotype-reference combination will be associated with all the genes. Checks performed on the data submitted via the curation interface ensure that all required values are supplied by the curator and that all submitted values, such as reference ID numbers, are valid.
User displays
SGD users view phenotype information on three types of SGD web page: the Locus Summary page for an individual gene, which includes a brief summary of associated mutant phenotypes; the Phenotype Details page for an individual gene, where complete phenotype information is listed; and Search Result pages, that aggregate shared phenotype information from different genes. A major consideration in designing the user interfaces was that users should be able to search and view phenotypes in a straightforward manner without having to understand all the details of this fairly complex curation system.
The Locus Summary page displays a subset of information for each phenotype: the high-level experiment type ‘Classical genetics’ or ‘Large-scale survey’; the mutant type; and the observable and qualifier (Figure 2). Because some observables use very general language, in order to provide more information at a glance for users we substitute additional phenotype properties in the phrase shown for those observables on the Locus Summary. For example, rather than displaying the observable and qualifier ‘resistance to chemicals: decreased’ on the Locus Summary, the name of the chemical is substituted within the phrase so that it is shown as, for instance, ‘resistance to cycloheximide: decreased’. An analogous substitution is made for particular observables that are used with the Reporter property: instead of displaying ‘protein/peptide modification: abnormal’ where the Reporter is carboxypeptidase Y, ‘carboxypeptidase Y (Prc1p) modification: abnormal’ is displayed, giving an immediate indication that the phenotype involves the vacuolar protein sorting pathway. Each observable-qualifier pair shown on the Locus Summary is hyperlinked to a list of all other mutant phenotype annotations containing that observable and qualifier, as well as the genes and references associated with them. Where a substitution has been made, the hyperlink leads to an observable–qualifier–property combination to accommodate the substituted text, such that for the example above, ‘resistance to cycloheximide: decreased’ is linked to a list of all other mutant phenotypes incorporating decreased resistance to cycloheximide, as opposed to decreased ‘resistance to chemicals’.

Phenotype section of SGD's Locus Summary page for the ALG8 gene. The figure represents only a portion of the SGD locus summary for ALG8, http://www.yeastgenome.org/cgi-bin/locus.fpl?locus=ALG8. The observable, qualifier, and high-level experiment type are shown for all curated mutant phenotypes of ALG8. For phenotypes that include the observable ‘resistance to chemicals’, the name of the chemical is substituted for the word ‘chemical’ in this summary display; several examples of this are shown here. For the observable ‘protein/peptide modification’, the name of the Reporter is substituted for ‘protein/peptide’—in this instance, ‘carboxypeptidase Y (Prc1p)’.
At the top of the Locus Summary, tabs lead to pages with additional detailed information about the gene. The ‘Phenotype’ tab leads to the Phenotype Details page, which displays a table of all mutant phenotypes that have been curated for that gene (Figure 3). Columns of the table include Experiment type; Mutant information (incorporating both Mutant type and Allele information, if any); Strain background; Phenotype (observable and qualifier); Chemical (any chemicals relevant to the phenotype); Details (including the properties ‘Details’, ‘Condition’, and ‘Reporter’), and Reference. Each row of the table contains a single phenotype from a single reference. Each observable in the table is hyperlinked to a list of all other phenotypes incorporating that observable, and any chemical names are linked to lists of all phenotypes involving that chemical. The page contains links to download all phenotype data for that gene in a tab-delimited file, and to browse the entire YPO, as well as links to external databases relevant to yeast phenotypes and to sources for yeast strains and plasmids. In addition, all web pages containing phenotype data have links to extensive user help documentation that conveys basic information about yeast phenotypes, the curation system, and use of the search interfaces.

Phenotype details page for the ALG8 gene. Only a portion of the table is shown. Columns of the table contain: experiment type; mutant type and allele information, if any; strain background; phenotype (observable: qualifier); chemical, if any; other details, including conditions, reporters, or details; and the reference. Each observable name in the phenotype column is hyperlinked to a list of other phenotype annotations using that observable. Each chemical name in the chemical column is hyperlinked to a list of other phenotype annotations involving that chemical. This table of phenotypes is on a page that contains the standard SGD toolbar containing links to major tools and resources in SGD; below the table are links to other resources, external to SGD, which offer mutant phenotype information or provide mutant strains (data not shown).
Search interfaces and results pages
Currently, SGD users may search phenotype data in two different ways (Figure 4). The basic SGD Search is accessed via a text box that appears at the top of most SGD web pages. This search accesses multiple different kinds of information in SGD, including gene names and descriptions, GO terms, phenotypes, biochemical pathways, and more. At present, the only type of phenotype data searched is the observables, as it would be too computationally intensive to have this basic search access all the different aspects of phenotype data (we plan to address this in the future, as described below). Search results (Figure 4) are presented as links to lists of phenotypes containing observables that match the search criterion.
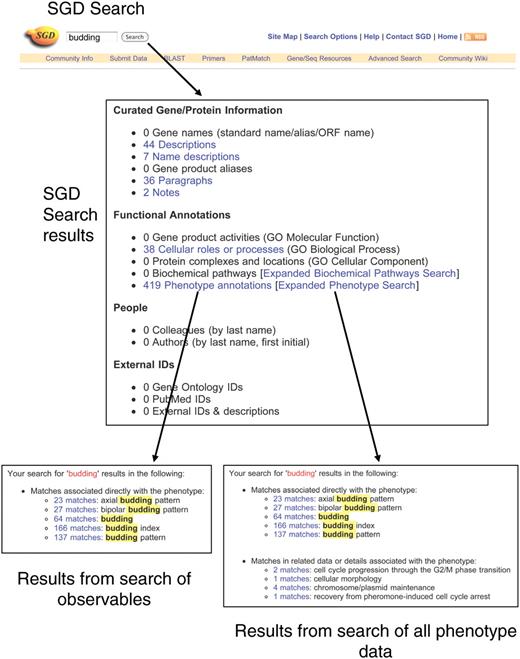
Searching phenotype data. The basic SGD Search is entered via a text box at the top of most SGD pages. Keywords entered into this search are used to search 15 major categories of information in SGD, and a summary of the results is presented on the SGD search results page (center). The link to ‘Phenotype annotations’ leads to a summary page listing the number of matches found in observables (lower left). The link to ‘Expanded phenotype search’ launches a search of all phenotype data. Results of the search are presented on the Expanded Phenotype Search Results Summary page (lower right). The top section displays matches to observables, and the bottom section displays matches to other phenotype data. On both types of results summary page, each number of matches is linked to a page displaying a table of the individual annotations.
A separate search tool, the Expanded Phenotype Search, scans the complete set of phenotype data, including properties such as conditions, alleles, strain backgrounds, or chemicals. The Expanded Phenotype Search tool can be accessed via a link on the basic search results page, or directly via a link on SGD's search options page (http://www.yeastgenome.org/SearchContents.shtml), which lists all the available tools for searching SGD data. The results page for the Expanded Phenotype search presents a list of phenotype annotations matching the search criterion, separated into two categories: matches to the observable and matches to other phenotype data.
To facilitate browsing of all phenotype information, the entire YPO hierarchy of observables is displayed on a web page on which each term is linked to the complete list of mutant phenotypes annotated using that observable (http://www.yeastgenome.org/cache/PhenotypeTree.html). This page displaying the YPO is linked from several different types of page in SGD that display phenotype data, so that it is conveniently accessible to anyone who is viewing or searching mutant phenotypes.
To provide users with a comprehensive and computer-readable overview of mutant phenotype data, we provide a file, ‘phenotype_data.tab’, on the SGD Download Data Files page (http://downloads.yeastgenome.org/) that contains all phenotype data in SGD and is updated weekly. Table 4 illustrates the format of the file, which, like all other SGD data, is freely available for download. SGD's Batch Download tool allows users to create a file in the same format containing phenotype data for a user-specified set of genes.
Format of the ‘phenotype_data.tab’ file available from SGD's download data files page (http://www.yeastgenome.org/cgi-bin/batchDownload/)
| Column number | Column name | Mandatory? | Column contents |
|---|---|---|---|
| 1 | Feature name | Yes | The systematic name of the gene |
| 2 | Feature type | Yes | The feature type of the gene |
| 3 | Gene name | No | The SGD Standard Name of the gene |
| 4 | SGDID | Yes | The SGDID, unique database identifier, for the gene |
| 5 | Reference | Yes | PubMed ID | SGD Reference Identifier |
| 6 | Experiment type | Yes | The method used to detect and analyze the phenotype |
| 7 | Mutant type | Yes | Description of the impact of the mutation on activity of the gene product |
| 8 | Allele | No | Allele name and description |
| 9 | Strain background | Noa | Genetic background in which the phenotype was analyzed |
| 10 | Phenotype | Yes | Observable: qualifier |
| 11 | Chemical | Nob | Any chemical relevant to the phenotype |
| 12 | Condition | No | Condition under which the phenotype is observed |
| 13 | Details | No | Details about the phenotype |
| 14 | Reporter | Noc | The protein(s) or RNA(s) used in an experiment to track a process |
| Column number | Column name | Mandatory? | Column contents |
|---|---|---|---|
| 1 | Feature name | Yes | The systematic name of the gene |
| 2 | Feature type | Yes | The feature type of the gene |
| 3 | Gene name | No | The SGD Standard Name of the gene |
| 4 | SGDID | Yes | The SGDID, unique database identifier, for the gene |
| 5 | Reference | Yes | PubMed ID | SGD Reference Identifier |
| 6 | Experiment type | Yes | The method used to detect and analyze the phenotype |
| 7 | Mutant type | Yes | Description of the impact of the mutation on activity of the gene product |
| 8 | Allele | No | Allele name and description |
| 9 | Strain background | Noa | Genetic background in which the phenotype was analyzed |
| 10 | Phenotype | Yes | Observable: qualifier |
| 11 | Chemical | Nob | Any chemical relevant to the phenotype |
| 12 | Condition | No | Condition under which the phenotype is observed |
| 13 | Details | No | Details about the phenotype |
| 14 | Reporter | Noc | The protein(s) or RNA(s) used in an experiment to track a process |
SGD's download data files page contains all phenotype data in SGD and is updated weekly. Its format is the same as the files created by the Batch Download tool (http://www.yeastgenome.org/cgi-bin/batchDownload), which may be used to retrieve phenotype and other data for a user-specified set of genes. aStrain background is mandatory in all current curation but was not entered for some of the first phenotypes curated. bA chemical is required for the following observables, because the name of the chemical is substituted for ‘chemical compound’ or ‘chemical’ on the Locus Summary page: chemical compound accumulation, chemical compound excretion, and resistance to chemicals. cA reporter is required for the following observables, because the name of the reporter is substituted for ‘protein’, ‘protein/peptide’ or ‘RNA’ on the Locus Summary page: protein activity, protein/peptide accumulation, protein/peptide distribution, protein/peptide modification, RNA accumulation, RNA localization, and RNA modification.
Format of the ‘phenotype_data.tab’ file available from SGD's download data files page (http://www.yeastgenome.org/cgi-bin/batchDownload/)
| Column number | Column name | Mandatory? | Column contents |
|---|---|---|---|
| 1 | Feature name | Yes | The systematic name of the gene |
| 2 | Feature type | Yes | The feature type of the gene |
| 3 | Gene name | No | The SGD Standard Name of the gene |
| 4 | SGDID | Yes | The SGDID, unique database identifier, for the gene |
| 5 | Reference | Yes | PubMed ID | SGD Reference Identifier |
| 6 | Experiment type | Yes | The method used to detect and analyze the phenotype |
| 7 | Mutant type | Yes | Description of the impact of the mutation on activity of the gene product |
| 8 | Allele | No | Allele name and description |
| 9 | Strain background | Noa | Genetic background in which the phenotype was analyzed |
| 10 | Phenotype | Yes | Observable: qualifier |
| 11 | Chemical | Nob | Any chemical relevant to the phenotype |
| 12 | Condition | No | Condition under which the phenotype is observed |
| 13 | Details | No | Details about the phenotype |
| 14 | Reporter | Noc | The protein(s) or RNA(s) used in an experiment to track a process |
| Column number | Column name | Mandatory? | Column contents |
|---|---|---|---|
| 1 | Feature name | Yes | The systematic name of the gene |
| 2 | Feature type | Yes | The feature type of the gene |
| 3 | Gene name | No | The SGD Standard Name of the gene |
| 4 | SGDID | Yes | The SGDID, unique database identifier, for the gene |
| 5 | Reference | Yes | PubMed ID | SGD Reference Identifier |
| 6 | Experiment type | Yes | The method used to detect and analyze the phenotype |
| 7 | Mutant type | Yes | Description of the impact of the mutation on activity of the gene product |
| 8 | Allele | No | Allele name and description |
| 9 | Strain background | Noa | Genetic background in which the phenotype was analyzed |
| 10 | Phenotype | Yes | Observable: qualifier |
| 11 | Chemical | Nob | Any chemical relevant to the phenotype |
| 12 | Condition | No | Condition under which the phenotype is observed |
| 13 | Details | No | Details about the phenotype |
| 14 | Reporter | Noc | The protein(s) or RNA(s) used in an experiment to track a process |
SGD's download data files page contains all phenotype data in SGD and is updated weekly. Its format is the same as the files created by the Batch Download tool (http://www.yeastgenome.org/cgi-bin/batchDownload), which may be used to retrieve phenotype and other data for a user-specified set of genes. aStrain background is mandatory in all current curation but was not entered for some of the first phenotypes curated. bA chemical is required for the following observables, because the name of the chemical is substituted for ‘chemical compound’ or ‘chemical’ on the Locus Summary page: chemical compound accumulation, chemical compound excretion, and resistance to chemicals. cA reporter is required for the following observables, because the name of the reporter is substituted for ‘protein’, ‘protein/peptide’ or ‘RNA’ on the Locus Summary page: protein activity, protein/peptide accumulation, protein/peptide distribution, protein/peptide modification, RNA accumulation, RNA localization, and RNA modification.
Future directions
Software improvements
Some yeast genes already have more than 50 curated phenotypes in SGD, and that number will continue to rise as more large-scale phenotype studies are published. As the data expand, it becomes increasingly necessary to sort and display them in versatile ways in order to view phenotypes of particular interest. Currently, the phenotype data may be downloaded to the user's computer and sorted in file format. Eventually we aim to display them in a sortable and filterable table on the web page, as is already in use for physical and genetic interaction data. Although the complexity of the phenotype data makes this challenging, it is an attainable goal for the near future. We are also investigating ways, such as using materialized views, in which the basic SGD search could comprehensively and quickly search all phenotype data, thus incorporating the Expanded Phenotype Search functionality and eliminating the need for two separate search tools.
Additional analysis tools
In the future we will develop search and analysis tools that exploit the hierarchical structure of the YPO observables and qualifiers, as well as experiment types. This task is greatly facilitated by the fact that such tools have already been developed for analysis of GO annotations.
A more sophisticated phenotype search tool will allow the user to start with a given observable, observable–qualifier combination, or experiment type, and retrieve genes annotated with those terms and any of their child terms. For example, one could search for ‘osmotic stress resistance’ and gather all genes annotated to that term as well as to the child terms ‘hyperosmotic stress resistance’ and ‘hypo-osmotic stress resistance’, or search for ‘filamentous growth: decreased’ and retrieve annotations to both ‘decreased’ and ‘absent’ filamentous growth. One could also restrict the search by experiment type, compiling results from the different types of large-scale study, or retrieving only annotations derived from small-scale classical genetics experiments. Additional phenotype data could also be included in SGD's Advanced Search tool. The search currently allows users to use the phenotypes ‘viable’ and ‘inviable’ as criteria, as well as selecting the type of chromosomal feature and annotation to a particular GO term or terms. Adding the ability to specify other phenotypes, in addition to ‘viable’ and ‘inviable’, will improve the utility of the search.
The GO Term Finder tool (13) compares the GO annotations for a list of genes specified by the user to those of a specified background set of genes, in order to calculate the significance of GO annotations shared among the input set. For example, a user might enter a list of genes that exhibit a similar expression profile in a microarray experiment, and find that many more of the genes are annotated to respiratory metabolism terms than would be expected by random chance; this would provide a clue that unknown genes in the set might also be involved in respiratory metabolism. Similarly, a Phenotype Term Finder would find significantly shared phenotypes among a group of genes, suggesting their common involvement in the corresponding process.
Subsets of high-level GO terms, called GO Slims, are useful for sorting gene products into broad categories according to their molecular function, subcellular localization, or the biological process in which they are involved (14). The GO Slim Mapper tool at SGD performs this sorting for a given list of genes and a GO Slim set of the user's choice (http://www.yeastgenome.org/SlimMapper). Although the YPO has many fewer terms than GO (about 160 terms as of October 2008, compared to about 2200 GO terms in the smallest GO aspect, Cellular Component), it will be useful to develop a very broad set of perhaps 20 observables representing general phenotypes, and to adapt the GO Slim Mapper as a Phenotype Slim Mapper tool.
Compatibility with other curation systems
Since the presently described curation system focuses on phenotypes that occur at the cellular level and, to some extent, at the molecular level, it can be easily extensible to other single-celled fungi. Other model organism databases have created phenotype ontologies tailored to the anatomy and physiology of those organisms, such as the Mammalian phenotype ontology, the C. elegans phenotype ontology, and the Cereal plant trait ontology (15–17). In contrast to the YPO, all three of these ontologies contain terms that combine the equivalents of the observable and qualifier, for example, ‘abnormal adipose tissue morphology’ in the Mouse phenotype ontology. There is some overlap between the YPO and these ontologies—for example, the Mammalian and C. elegans phenotype ontologies and the YPO all contain terms describing mitochondrial morphology—but the ontologies for multicellular organisms necessarily focus on the observable features of the organism rather than the cell.
During the time that we were developing this phenotype curation system, an effort was underway to create a phenotypic attribute trait ontology (PATO), that would be applicable to multiple organisms (see http://www.bioontology.org/wiki/index.php/PATO:Main_Page) (18). PATO is an ontology of ‘phenotypic qualities’ that is meant to be used in conjunction with other ontologies of ‘quality-bearing entities’, such as the YPO observables. PATO contains terms that are directly comparable to SGD's qualifiers, such as ‘abnormal’ and ‘decreased rate’, as well as many other terms that are not applicable to single-celled organisms. Some model organism databases, such as the Zebrafish Information Network (ZFIN; http://zfin.org), are actively using PATO terms for mutant phenotype curation (18). We chose not to incorporate PATO into our curation system because the PATO system was changing greatly while we were designing SGD's system. Further, PATO is best used to capture phenotypes of multicellular organisms. However, both systems have been evolving in a convergent manner, and it is now likely straightforward to map PATO terms to YPO qualifiers, and/or to apply any relevant PATO term to a YPO observable. Thus we are in a good position to integrate SGD's mutant phenotype curation into any larger effort that uses PATO terms.
Conclusions
Our aim in developing this mutant phenotype curation system has been to ensure that the curated data are readily accessible and understandable, for yeast researchers and SGD curators alike. We have tried to keep the components of the system simple, controlling vocabulary wherever possible and curating broader phenotypic characterizations that can be grouped for searching purposes. We also built in enough flexibility in the entry of free text that any phrases or concepts that are commonly used by researchers, but may not be included in the controlled vocabularies, can be incorporated into the curation. Having used the curation system to record phenotypes for nearly all yeast genes, we can conclude that the system is successful. The vocabularies it comprises will continue to evolve as new methods become available to assay yeast phenotypes.
Funding
US National Human Genome Research Institute (HG001315 to J.M.C.).
Conflict of interest. None declared.
Acknowledgements
We thank members of SGD's Scientific Advisory Board and our Stanford colleagues, in particular Gavin Sherlock and Jared Wenger, for helpful comments during the development of the curation system.
The SGD Project includes: Rama Balakrishnan, Gail Binkley, J. Michael Cherry, Karen R. Christie, Maria C. Costanzo, Selina Dwight, Stacia R. Engel, Dianna G. Fisk, Jodi E. Hirschman, Benjamin Hitz, Eurie L. Hong, Cynthia Krieger, Stuart Miyasato, Robert Nash, Julie Park, Marek S. Skrzypek, Shuai Weng, and Edith Wong (Stanford); David Botstein, Kara Dolinski, Mike Livstone, and Rose Oughtred (Princeton).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Comments
Note from the authors: Since this manuscript was prepared, the scope of the YPO has been expanded to include Aspergillus mutant phenotypes. The ontology of observables, qualifiers, and experiment types is now called the Ascomycete Phenotype Ontology (APO), and the master file, termed 'ascomycete_phenotype.obo', is available at the Open Biomedical Ontologies website, http://www.obofoundry.org/. Within the file, terms are grouped into overlapping species-specific subsets. This allows the extraction of a file containing only the terms relevant to a particular species, so that database users and curators need not view irrelevant terms.
Conflict of Interest:
None declared