





Abstract
Data and metadata interoperability between data storage systems is a critical component of the FAIR data principles. Programmatic and consistent means of reconciling metadata models between databases promote data exchange and thus increases its access to the scientific community. This process requires (i) metadata mapping between the models and (ii) software to perform the mapping. Here, we describe our efforts to map metadata associated with genome assemblies between the National Center for Biotechnology Information (NCBI) data resources and the Chado biological database schema. We present mappings for multiple NCBI data structures and introduce a Tripal software module, Tripal EUtils, to pull metadata from NCBI into a Tripal/Chado database. We discuss potential mapping challenges and solutions and provide suggestions for future development to further increase interoperability between these platforms.
Database URL: https://github.com/NAL-i5K/tripal_eutils
Introduction
Background
Biologists increasingly recognize the need to make data and metadata more findable, accessible, interoperable and reusable (FAIR) (1). These guiding principles provide a framework to guide the improvement of research data cyberinfrastructure and equip scientists to use public data to enhance knowledge discovery. Metadata—essentially, data describing the data—are an essential component of the FAIR data principles, as data without metadata cannot be reused. FAIR relies on data integration from different resources, a process that can be incredibly demanding as many datasets encompass different data types with complex connections. Modeling the full structure of data and metadata and creating appropriate linkages between datasets require sophisticated data storage structures, such as a relational database. When data exist in two different structures—whether these are flat files, relational databases or something else—an inability to map between those structures can slow down or entirely prevent data integration and thus data reuse.
The National Center for Biotechnology Information (NCBI) is the primary US data repository for nucleotide and protein datasets (2). NCBI houses numerous types of biological data, and where it is supported, researchers are encouraged or even required to submit their data to NCBI prior to publication. As one of the three databases in the International Nucleotide Sequence Database Collaboration (INSDC), it is often the first or only database where researchers deposit their data in. As such, it is the central warehouse for genetic and genomic data and their metadata in the life sciences. Data and metadata relevant to genetics and genomics are stored in at least seven data resources at NCBI (Assembly, BioProject, BioSample, PubMed, NCBITaxon, Genome and Nucleotide), underscoring how complex the data storage can be.
Community databases, on the other hand, provide value-added tools and services for smaller communities, for example those around a particular taxonomic group (3,4). These databases provide tools for data visualization, search and browse; store specialty data types; perform manual data curation and integration; serve as hubs for community discussion; and are often a liaison between biologists and INSDC databases—all invaluable services for research groups lacking dedicated bioinformatic support. The open-source biological database toolkit Tripal serves as a common framework for community databases (5). One of the major strengths of the Tripal framework is FAIR treatment of data and metadata. In particular, Tripal requires mapping of all content types, and all their associated metadata, to ontology terms. Tripal has become a well-adopted software for community databases, with at least 17 databases using the software in some capacity as of February 2019 (https://fairsharing.org/collection/Tripal). As such, Tripal represents numerous community hubs that can benefit from commonly needed solutions for biological data storage.
Integrating metadata across NCBI and community databases
Both community databases and NCBI can and should have access to the metadata records—preferably the same ones—that describe shared hosted datasets. Ideally, metadata exchange would happen programmatically to enable synchronization of metadata between resources—a tenet of the ‘interoperable’ principle of the FAIR data principles. For example, a scientist would submit metadata describing a genome sequencing and assembly project to NCBI in order to create entries in NCBI’s BioProject, BioSample and Assembly databases—and the community database would be able to programmatically extract the genome project’s metadata from NCBI and store it in a consistent, ontology-driven manner in the Chado database schema (6). That way, the scientist does not have to provide the metadata twice—to NCBI and the community database—reducing the burden on the scientist and the possibility of inadvertently providing inconsistent metadata between databases. Putting this idea successfully into practice requires (i) exposing relevant metadata in the ‘source’ database via web services, (ii) mapping a correspondence of the metadata model between databases and (iii) developing software to pull the metadata into the receiving database. NCBI offers a web services suite, E-utilities (Entrez Programming Utilities) (7), that enables programmatic retrieval of genome assembly-relevant metadata. However, the latter two requirements have not yet been developed for genome assembly-relevant metadata in Chado.
Tripal supports the community database schema standard Chado (6,8). Chado is the biological database schema endorsed by the generic model organism database (GMOD) community and is used by many community databases and software packages. Chado is a highly normalized relational database, with modules grouping tables of similar content. Certain tables, such as controlled vocabulary (CV) term or property tables, serve as linker or annotation tables and are implemented in a consistent way across Chado.
Historically, Chado was designed to be flexible to accommodate the diversity of use cases that adopting databases would encounter; for example, the model organism database FlyBase (9) is likely to have different needs than databases such as the Hardwood Genomics and i5k Workspace@NAL, which serve many smaller scientific communities, even though all databases store similar data types. However, this flexibility leads to different groups storing the same data and metadata types differently. Although some guidelines exist, these do not cover all data types and do not include metadata. For example, the community of databases using Chado store GO term annotations (10) for features in different Chado tables (feature_cvterm, featureprop or feature_dbxref), primarily due to lack of guidelines (https://github.com/GMOD/Chado/issues/74). When databases vary the way they store the same data types in Chado, it is difficult or impossible to share software. This is particularly relevant for Tripal-based site developers, who generally adopt the Tripal software in order to save on development costs and benefit from a standardized platform, and therefore would not want to develop their own data exchange software from scratch. To our knowledge, there are no community-developed guidelines for storing genome sequence and genome assembly metadata in Chado and, specifically, no suggested way to map the metadata from NCBI’s model to Chado tables.
The Tripal toolkit, with its native data loaders and display fields, is an excellent way to ensure data and metadata are stored in a compatible (if not identical) manner across community databases. However, Tripal does not yet provide importers for most assembly-level metadata. Currently available loaders focus on importing biological data (eg. GFF and FASTA files) rather than the associated metadata concerning generation of that data. There are no other definitive guidelines for how to load NCBI-structured metadata into Chado. We therefore sought to create a metadata map for genome assembly-relevant metadata between NCBI and Chado—metadata that should be relevant to most community databases that host genomic data. In order to facilitate adoption of this mapping, we present a Tripal extension module, Tripal EUtils, which accesses metadata from the NCBI Assembly, BioProject and BioSample resources using NCBI’s E-utilities and imports it to Chado using the proposed map. This module is available for installation on any Tripal-v3-based site.
Metadata mapping
In order to introduce our solution for translating from NCBI to Chado, we first must understand some key concepts in each system’s data model. NCBI’s data and metadata are partitioned into many individual databases, or resources (2), each respectively containing a primary data type, metadata and ancillary information (e.g. nucleotide data and metadata is stored in the ‘Nucleotide’ database). NCBI’s E-utilities are a set of server-side programs that interface with these resources (7). They allow a user or program to programmatically extract data and metadata from individual NCBI resources. The returned information, often in eXtensible Markup Language (XML) or JavaScript Object Notation format, is typically structured in attribute-value pairs, although more complex structures can be returned. These can be parsed by the requesting program as needed for ingest into a local database.
The Chado database consists of ‘modules’—groups of tables that store data and metadata of the same type. For example, sequence data are stored in the tables in the Sequence Module. Each module contains one or more ‘base’ tables that contain primary data for individual records. The organism base table, for example, stores information describing a biological species in each record. Ancillary data, such as properties or CV terms, are associated to a base record through ‘term’, ‘property’ and other related tables and connected to other base tables (such as the feature table containing genomic features) through linker tables.
Here, we describe the mapping of the NCBI E-utilities response object to Chado base, ancillary and linker tables. The Chado version used here is 1.3 and the Tripal version is 7.x-3.1. When additional custom tables or schema modifications are necessary, they are noted in the text. However, all suggested changes are backwards compatible to previous Chado versions. Additionally, this manuscript describes the mapping of NCBI attributes to community-defined controlled vocabularies. For this work, the European Bioinformatics Institute (EBI) ontology lookup service was used to find appropriate ontology terms. Preference was given to the well-established Semanticscience Integrated Ontology (SIO) (11), Experimental Factor Ontology (EFO) (12) and EDAM (12) ontologies wherever possible. To demonstrate cross-species utility of our mapping approach, the following set of records from insects, mammals and plants were used to develop and test NCBI–Chado metadata mappings: assembly uids 1949871 (honey bee), 317138 (dog), 524058 (yak), 557018 (wild strawberry), 751381 (rubber tree), 2004951 (hemp), 559011 (English walnut), 91111 (locust) and their corresponding BioProjects and BioSamples.
Goals and rules for mapping
The following principles guided our mappings from NCBI to Chado. First, we only mapped metadata that are relevant to a genome assembly (as opposed to NCBI-specific data, such as internal identifiers). Second, we sought to avoid changes to the Chado schema. Where we do suggest new Chado tables, we do so only when changes are necessary for Tripal EUtils, do not break backwards compatibility and reasonably meet a need within the Chado and Tripal communities beyond the use case of this module. Third, in some rare cases, the property or content may be already mapped in the main Tripal codebase. In this event, that mapping was preferred. Finally, mapping records 1:1 between NCBI and Chado is preferred, as splitting a single NCBI record (which corresponds to a single unique ID) across multiple records in Chado (individual entries in a table) results in difficult questions of where to map corresponding metadata. For example, if we need to decide whether an NCBI record should be split into multiple Chado records, or retained as one Chado record, we will default to the latter.
Attribute names, or tags, returned by NCBI’s E-utilities service are not derived from a published CV or ontology. To our knowledge, there is no comprehensive list, CV or other documentation of attribute tags for Assembly metadata that our mappings could rely on. Therefore, it was necessary to make assumptions about certain mappings between NCBI and Chado. We note that this approach is problematic because returned XML tags could change without notice. We contrast this to metadata returned via the Attributes XML tag from NCBI’s BioSample resource. The value of this tag holds the relevant metadata for a variety of properties from a CV (https://www.ncbi.nlm.nih.gov/biosample/docs/packages/?format=xml), greatly reducing the difficulty in identifying the nature of the metadata and therefore its analogous mapping in Chado.
NCBI’s metadata resources for genome assemblies
Before defining mappings between NCBI and Chado, we must identify the relevant database resources in NCBI for this project. There are at least 38 distinct database resources in NCBI (2), yet only a subset of these are relevant to the genomic-centric databases we develop for. As discussed in the previous section, we focus on NCBI databases that (i) make their data and metadata available via NCBI’s E-utilities and (ii) represent data and/or metadata relevant to genome assemblies. This covers the collection of isolates, sequencing and assembly of genomes: NCBI does not currently house metadata for the generation of annotations, a role filled by the community databases. Therefore, we limited our mapping to the NCBI resources Assembly, BioProject, BioSample, PubMed and NCBITaxon. NCBI’s Genome resource provides metadata that mostly overlaps with other NCBI resources. Data and metadata from the nucleotide resource would typically be ingested into Chado by other sources, such as via a GFF3 file.
Proposed mappings
The following sections describe our proposal for mapping NCBI’s Assembly, BioProject, BioSample, PubMed and NCBITaxon into Chado. Moreover, it also describes the implementation of that mapping performed by the Tripal EUtils module.
Mapping NCBI’s BioProject content
NCBI defines BioProjects as ‘... a collection of biological data related to a single initiative, originating from a single organization or from a consortium. A BioProject record provides users a single place to find links to the diverse data types generated for that project’ (13). Chado has a project table; the schema description is ‘Standard Chado flexible property table for projects’. The table has a name and description field only; Chado 1.4 will introduce a type_id column to the project table. We propose mapping NCBI’s BioProject resource to the Chado Project table (Table 1). Unfortunately, no clear term for the NCBI Project exists in a generic ontology or CV; we therefore do not propose a type column.
Mappings for BioProject metadata returned from NCBI’s E-utilities into Chado
| NCBI XML | Chado base table | Chado column |
|---|---|---|
| ProjectDescr- > Name and ProjectDescr- > Title | project | name |
| ProjectDescr- > Description | project | description |
| ProjectID | dbxref | project_dbxref |
| NCBI XML | Chado base table | Chado column |
|---|---|---|
| ProjectDescr- > Name and ProjectDescr- > Title | project | name |
| ProjectDescr- > Description | project | description |
| ProjectID | dbxref | project_dbxref |
The NCBI XML column contains tags from the XML-formatted content returned by E-utilities.
Hierarchical content is represented by an arrow, where the term to the left of the arrow is the parent term and that to the right of the arrow is the child term.
Chado base table refers to the main table the content is mapped to in Chado; Chado column describes where in the table the tag will be mapped.
Mappings for BioProject metadata returned from NCBI’s E-utilities into Chado
| NCBI XML | Chado base table | Chado column |
|---|---|---|
| ProjectDescr- > Name and ProjectDescr- > Title | project | name |
| ProjectDescr- > Description | project | description |
| ProjectID | dbxref | project_dbxref |
| NCBI XML | Chado base table | Chado column |
|---|---|---|
| ProjectDescr- > Name and ProjectDescr- > Title | project | name |
| ProjectDescr- > Description | project | description |
| ProjectID | dbxref | project_dbxref |
The NCBI XML column contains tags from the XML-formatted content returned by E-utilities.
Hierarchical content is represented by an arrow, where the term to the left of the arrow is the parent term and that to the right of the arrow is the child term.
Chado base table refers to the main table the content is mapped to in Chado; Chado column describes where in the table the tag will be mapped.
Mapping NCBI’s Assembly content
The NCBI’s Assembly database resource provides information on assembled genomes, including structure, names, identifiers, statistics, assembly history and cross-references to other databases (14). We propose to store the assembly metadata in the Chado analysis table module (Table 2). The analysis table is defined in Chado very particularly: ‘An analysis is a particular type of computational analysis; it may be a blast of one sequence against another, or an all by all blast, or a different kind of analysis altogether. It is a single unit of computation.’ (http://gmod.org/wiki/Chado_Companalysis_Module#Table:_analysis). Note that storing the genome assembly metadata in the analysis table requires us to refer to a genome assembly as ‘a single unit of computation’.
Proposed mappings for Assembly metadata from NCBI’s E-utilities into Chado
| NCBI XML | Chado base table | Chado column |
|---|---|---|
| AssemblyName | analysis | name |
| AssemblyDescription | analysis | description |
| # Assembly method: (from FTP) | analysis | program |
| # Assembly method: (from FTP) | analysis | programversion |
| N/A | analysis | algorithm |
| BioSampleAccn | analysis | sourcename |
| N/A | analysis | sourceversion |
| N/A | analysis | sourceuri |
| SubmissionDate | analysis | timeexecuted |
| Stats | analysisprop | type/value |
| FtpSites | analysisprop | type/value |
| RsUid | dbxref | accession |
| GbUid | dbxref | accession |
| NCBI XML | Chado base table | Chado column |
|---|---|---|
| AssemblyName | analysis | name |
| AssemblyDescription | analysis | description |
| # Assembly method: (from FTP) | analysis | program |
| # Assembly method: (from FTP) | analysis | programversion |
| N/A | analysis | algorithm |
| BioSampleAccn | analysis | sourcename |
| N/A | analysis | sourceversion |
| N/A | analysis | sourceuri |
| SubmissionDate | analysis | timeexecuted |
| Stats | analysisprop | type/value |
| FtpSites | analysisprop | type/value |
| RsUid | dbxref | accession |
| GbUid | dbxref | accession |
The NCBI XML column contains tags from the XML-formatted content returned by NCBI’s E-utilities.
Hierarchical content is represented by an arrow, where the term to the left of the arrow is the parent term and that to the right of the arrow is the child term.
Chado base table refers to the main table the content is mapped to in Chado; Chado column describes where in the table the tag will be mapped.
N/A is used when the Chado base table contains non-required columns that are not provided in the metadata derived from NCBI’s E-utilities.
Proposed mappings for Assembly metadata from NCBI’s E-utilities into Chado
| NCBI XML | Chado base table | Chado column |
|---|---|---|
| AssemblyName | analysis | name |
| AssemblyDescription | analysis | description |
| # Assembly method: (from FTP) | analysis | program |
| # Assembly method: (from FTP) | analysis | programversion |
| N/A | analysis | algorithm |
| BioSampleAccn | analysis | sourcename |
| N/A | analysis | sourceversion |
| N/A | analysis | sourceuri |
| SubmissionDate | analysis | timeexecuted |
| Stats | analysisprop | type/value |
| FtpSites | analysisprop | type/value |
| RsUid | dbxref | accession |
| GbUid | dbxref | accession |
| NCBI XML | Chado base table | Chado column |
|---|---|---|
| AssemblyName | analysis | name |
| AssemblyDescription | analysis | description |
| # Assembly method: (from FTP) | analysis | program |
| # Assembly method: (from FTP) | analysis | programversion |
| N/A | analysis | algorithm |
| BioSampleAccn | analysis | sourcename |
| N/A | analysis | sourceversion |
| N/A | analysis | sourceuri |
| SubmissionDate | analysis | timeexecuted |
| Stats | analysisprop | type/value |
| FtpSites | analysisprop | type/value |
| RsUid | dbxref | accession |
| GbUid | dbxref | accession |
The NCBI XML column contains tags from the XML-formatted content returned by NCBI’s E-utilities.
Hierarchical content is represented by an arrow, where the term to the left of the arrow is the parent term and that to the right of the arrow is the child term.
Chado base table refers to the main table the content is mapped to in Chado; Chado column describes where in the table the tag will be mapped.
N/A is used when the Chado base table contains non-required columns that are not provided in the metadata derived from NCBI’s E-utilities.
A possible problem storing assembly metadata in the analysis table is that a genome assembly is generally multiple units of computation: multiple assembly algorithms may be used, scaffolding performed, etc. Therefore, we could have suggested storing an assembly in the Chado project table. To do this, we would generate a project record representing the assembly and then create individual analysis records for each program run to generate the assembly. While this may seem straightforward, two required fields for the Chado analysis table, program and programversion, are challenging to retrieve from NCBI. They are not returned by the XML. Instead, this information must be retrieved from an assembly summary file, the URL of which is given in the XML. Tripal EUtils downloads this file via FTP and parses the ‘# Assembly method’ line to retrieve a string containing program name(s) and version(s). To parse this string into separate program and their version, we would have to make additional assumptions about how the string is generated. Subsequently, metadata parsed from the NCBI Assembly record would then be split among multiple records within the analysis table, as well as the project table, creating a complex set of related metadata. For these reasons, we chose not to map the assembly metadata to the project and analysis tables.
Mapping NCBI’s BioSample content
The NCBI BioSample resource maps to Chado’s Biomaterial table (Table 3). Chado defines biomaterials in this manner, ‘A biomaterial represents the MAGE concept of BioSource, BioSample, and LabeledExtract. It is essentially some biological material (tissue, cells, serum) that may have been processed. Processed biomaterials should be traceable back to raw biomaterials via the biomaterialrelationship table.’ (http://gmod.org/wiki/Chado_Mage_Module#Table:_biomaterial).
Proposed mappings for NCBI BioSample into the Chado biomaterial table
| NCBI XML | Chado base table | Chado column |
|---|---|---|
| BioSample- > accession | biomaterial | name |
| Owner- > Name | contact | biomaterial.biosourceprovider_id |
| Comment- > Paragraph | biomaterial | description |
| Attribute | biomaterialprop | type_id, value |
| Organism- > taxonomy_id | organism_dbxref | dbxref.accession |
| NCBI XML | Chado base table | Chado column |
|---|---|---|
| BioSample- > accession | biomaterial | name |
| Owner- > Name | contact | biomaterial.biosourceprovider_id |
| Comment- > Paragraph | biomaterial | description |
| Attribute | biomaterialprop | type_id, value |
| Organism- > taxonomy_id | organism_dbxref | dbxref.accession |
In some cases, the metadata will be distributed across multiple tables; in this case, the value in the Chado column will contain the final table and column, separated by a period, in which the value of the record resides (e.g. dbxref.accession).
Proposed mappings for NCBI BioSample into the Chado biomaterial table
| NCBI XML | Chado base table | Chado column |
|---|---|---|
| BioSample- > accession | biomaterial | name |
| Owner- > Name | contact | biomaterial.biosourceprovider_id |
| Comment- > Paragraph | biomaterial | description |
| Attribute | biomaterialprop | type_id, value |
| Organism- > taxonomy_id | organism_dbxref | dbxref.accession |
| NCBI XML | Chado base table | Chado column |
|---|---|---|
| BioSample- > accession | biomaterial | name |
| Owner- > Name | contact | biomaterial.biosourceprovider_id |
| Comment- > Paragraph | biomaterial | description |
| Attribute | biomaterialprop | type_id, value |
| Organism- > taxonomy_id | organism_dbxref | dbxref.accession |
In some cases, the metadata will be distributed across multiple tables; in this case, the value in the Chado column will contain the final table and column, separated by a period, in which the value of the record resides (e.g. dbxref.accession).
A core component of NCBI’s BioSample metadata are the BioSample ‘packages’ (https://www.ncbi.nlm.nih.gov/biosample/docs/packages/). Data submitters can choose a package, which contains a variety of attribute sets such as plant- or insect-specific attributes, attribute values as recommended by the MIxS standard (15), etc. Here, we provide suggested ontology term mappings for attributes from the Invertebrate 1.0 and Plant 1.0 packages (https://www.ncbi.nlm.nih.gov/biosample/docs/packages/Invertebrate.1.0/, NCBI XML provided as Supplementary File S1; https://www.ncbi.nlm.nih.gov/biosample/docs/packages/Plant.1.0/, NCBI XML provided as Supplementary File S2; Table 4). The Chado property linking tables biomaterialprop, organismprop, analysisprop and projectprop are designed to hold such key:value pairs, and each attribute key must be associated to a vocabulary term housed in Chado’s cvterm table. Unfortunately, these NCBI attribute keys do not include semantic information like a true ontology and are not easily searchable using services like the EBI ontology lookup service (16) and it is not clear whether they are versioned. Ideally, we would implement these mappings in the Tripal EUtils software described in this paper (see below). However, if NCBI’s term names change, we would need to create new mappings. In the future, we aim to work with NCBI to include ontology definitions with the attributes themselves than to map them on the Tripal side only. In the meantime, instead of implementing the mappings in Table 4 via the Tripal EUtils software, we associate these attributes with a local vocabulary in Chado.
Proposed property mappings from the NCBI BioSample attribute packages for invertebrates and plants to ontology terms
| Attribute | Proposed ontology term(s) name | Proposed ontology term (accession) | Plant package | Invertebrate package |
|---|---|---|---|---|
| age | age | SIO:001013 | TRUE | TRUE |
| altitude | altitude | SIO:000438 | FALSE | TRUE |
| biomaterial_provider | biomaterial provider | EFO:0001729 | TRUE | TRUE |
| bioproject_accession | N/A | N/A | TRUE | TRUE |
| breed | breed | EFO:0005238 | FALSE | TRUE |
| cell_line | cell line | SIO:010054 | TRUE | FALSE |
| cell_type | cell type | EFO:0000324 | TRUE | FALSE |
| collected_by | TRUE | TRUE | ||
| collection_date | TRUE | TRUE | ||
| cultivar | cultivar | EFO:0005136 | TRUE | FALSE |
| culture_collection | TRUE | FALSE | ||
| depth | depth | SIO:000039 | FALSE | TRUE |
| description | description | schema:description | TRUE | TRUE |
| dev_stage | developmental stage | EFO:0000399 | TRUE | TRUE |
| disease | disease | SIO:010299 | TRUE | FALSE |
| disease_stage | disease stage | OBI:0000278 | TRUE | FALSE |
| ecotype | ecotype | EFO:0000434 | TRUE | FALSE |
| env_broad_scale | biome | ENVO:00000428 | FALSE | TRUE |
| genotype | genotype | SIO:001079 | TRUE | FALSE |
| geo_loc_name | geographic location | data:3720 | TRUE | TRUE |
| growth_protocol | growth protocol | EFO:0003789 | TRUE | FALSE |
| height_or_length | height/length | SIO:000040 height, SIO:000041 length | TRUE | FALSE |
| host | host | SIO:010415 | FALSE | TRUE |
| host_tissue_sampled | FALSE | TRUE | ||
| identified_by | FALSE | TRUE | ||
| isolate | TRUE | TRUE | ||
| isolation_source | Isolation source | data:3721 | TRUE | TRUE |
| lat_lon | Longitude, latitude | SIO:000318, SIO:000319 | TRUE | TRUE |
| organism | organism | OBI:0100026 | TRUE | TRUE |
| phenotype | phenotype | SIO:010056 | TRUE | FALSE |
| population | population | SIO:001061 | TRUE | FALSE |
| sample_name | name | schema:name | TRUE | TRUE |
| sample_title | title | SIO:000185 | TRUE | TRUE |
| sample_type | rdfs:type | TRUE | FALSE | |
| sex | biological sex | SIO:010029 | TRUE | TRUE |
| specimen_voucher | TRUE | TRUE | ||
| temp | temperature | EFO:0001702 | TRUE | TRUE |
| tissue | tissue | SIO:010002 | TRUE | TRUE |
| treatment | treatment | EFO:0000727 | TRUE | FALSE |
| Attribute | Proposed ontology term(s) name | Proposed ontology term (accession) | Plant package | Invertebrate package |
|---|---|---|---|---|
| age | age | SIO:001013 | TRUE | TRUE |
| altitude | altitude | SIO:000438 | FALSE | TRUE |
| biomaterial_provider | biomaterial provider | EFO:0001729 | TRUE | TRUE |
| bioproject_accession | N/A | N/A | TRUE | TRUE |
| breed | breed | EFO:0005238 | FALSE | TRUE |
| cell_line | cell line | SIO:010054 | TRUE | FALSE |
| cell_type | cell type | EFO:0000324 | TRUE | FALSE |
| collected_by | TRUE | TRUE | ||
| collection_date | TRUE | TRUE | ||
| cultivar | cultivar | EFO:0005136 | TRUE | FALSE |
| culture_collection | TRUE | FALSE | ||
| depth | depth | SIO:000039 | FALSE | TRUE |
| description | description | schema:description | TRUE | TRUE |
| dev_stage | developmental stage | EFO:0000399 | TRUE | TRUE |
| disease | disease | SIO:010299 | TRUE | FALSE |
| disease_stage | disease stage | OBI:0000278 | TRUE | FALSE |
| ecotype | ecotype | EFO:0000434 | TRUE | FALSE |
| env_broad_scale | biome | ENVO:00000428 | FALSE | TRUE |
| genotype | genotype | SIO:001079 | TRUE | FALSE |
| geo_loc_name | geographic location | data:3720 | TRUE | TRUE |
| growth_protocol | growth protocol | EFO:0003789 | TRUE | FALSE |
| height_or_length | height/length | SIO:000040 height, SIO:000041 length | TRUE | FALSE |
| host | host | SIO:010415 | FALSE | TRUE |
| host_tissue_sampled | FALSE | TRUE | ||
| identified_by | FALSE | TRUE | ||
| isolate | TRUE | TRUE | ||
| isolation_source | Isolation source | data:3721 | TRUE | TRUE |
| lat_lon | Longitude, latitude | SIO:000318, SIO:000319 | TRUE | TRUE |
| organism | organism | OBI:0100026 | TRUE | TRUE |
| phenotype | phenotype | SIO:010056 | TRUE | FALSE |
| population | population | SIO:001061 | TRUE | FALSE |
| sample_name | name | schema:name | TRUE | TRUE |
| sample_title | title | SIO:000185 | TRUE | TRUE |
| sample_type | rdfs:type | TRUE | FALSE | |
| sex | biological sex | SIO:010029 | TRUE | TRUE |
| specimen_voucher | TRUE | TRUE | ||
| temp | temperature | EFO:0001702 | TRUE | TRUE |
| tissue | tissue | SIO:010002 | TRUE | TRUE |
| treatment | treatment | EFO:0000727 | TRUE | FALSE |
Where no ontology terms were found, fields are left blank.
Proposed property mappings from the NCBI BioSample attribute packages for invertebrates and plants to ontology terms
| Attribute | Proposed ontology term(s) name | Proposed ontology term (accession) | Plant package | Invertebrate package |
|---|---|---|---|---|
| age | age | SIO:001013 | TRUE | TRUE |
| altitude | altitude | SIO:000438 | FALSE | TRUE |
| biomaterial_provider | biomaterial provider | EFO:0001729 | TRUE | TRUE |
| bioproject_accession | N/A | N/A | TRUE | TRUE |
| breed | breed | EFO:0005238 | FALSE | TRUE |
| cell_line | cell line | SIO:010054 | TRUE | FALSE |
| cell_type | cell type | EFO:0000324 | TRUE | FALSE |
| collected_by | TRUE | TRUE | ||
| collection_date | TRUE | TRUE | ||
| cultivar | cultivar | EFO:0005136 | TRUE | FALSE |
| culture_collection | TRUE | FALSE | ||
| depth | depth | SIO:000039 | FALSE | TRUE |
| description | description | schema:description | TRUE | TRUE |
| dev_stage | developmental stage | EFO:0000399 | TRUE | TRUE |
| disease | disease | SIO:010299 | TRUE | FALSE |
| disease_stage | disease stage | OBI:0000278 | TRUE | FALSE |
| ecotype | ecotype | EFO:0000434 | TRUE | FALSE |
| env_broad_scale | biome | ENVO:00000428 | FALSE | TRUE |
| genotype | genotype | SIO:001079 | TRUE | FALSE |
| geo_loc_name | geographic location | data:3720 | TRUE | TRUE |
| growth_protocol | growth protocol | EFO:0003789 | TRUE | FALSE |
| height_or_length | height/length | SIO:000040 height, SIO:000041 length | TRUE | FALSE |
| host | host | SIO:010415 | FALSE | TRUE |
| host_tissue_sampled | FALSE | TRUE | ||
| identified_by | FALSE | TRUE | ||
| isolate | TRUE | TRUE | ||
| isolation_source | Isolation source | data:3721 | TRUE | TRUE |
| lat_lon | Longitude, latitude | SIO:000318, SIO:000319 | TRUE | TRUE |
| organism | organism | OBI:0100026 | TRUE | TRUE |
| phenotype | phenotype | SIO:010056 | TRUE | FALSE |
| population | population | SIO:001061 | TRUE | FALSE |
| sample_name | name | schema:name | TRUE | TRUE |
| sample_title | title | SIO:000185 | TRUE | TRUE |
| sample_type | rdfs:type | TRUE | FALSE | |
| sex | biological sex | SIO:010029 | TRUE | TRUE |
| specimen_voucher | TRUE | TRUE | ||
| temp | temperature | EFO:0001702 | TRUE | TRUE |
| tissue | tissue | SIO:010002 | TRUE | TRUE |
| treatment | treatment | EFO:0000727 | TRUE | FALSE |
| Attribute | Proposed ontology term(s) name | Proposed ontology term (accession) | Plant package | Invertebrate package |
|---|---|---|---|---|
| age | age | SIO:001013 | TRUE | TRUE |
| altitude | altitude | SIO:000438 | FALSE | TRUE |
| biomaterial_provider | biomaterial provider | EFO:0001729 | TRUE | TRUE |
| bioproject_accession | N/A | N/A | TRUE | TRUE |
| breed | breed | EFO:0005238 | FALSE | TRUE |
| cell_line | cell line | SIO:010054 | TRUE | FALSE |
| cell_type | cell type | EFO:0000324 | TRUE | FALSE |
| collected_by | TRUE | TRUE | ||
| collection_date | TRUE | TRUE | ||
| cultivar | cultivar | EFO:0005136 | TRUE | FALSE |
| culture_collection | TRUE | FALSE | ||
| depth | depth | SIO:000039 | FALSE | TRUE |
| description | description | schema:description | TRUE | TRUE |
| dev_stage | developmental stage | EFO:0000399 | TRUE | TRUE |
| disease | disease | SIO:010299 | TRUE | FALSE |
| disease_stage | disease stage | OBI:0000278 | TRUE | FALSE |
| ecotype | ecotype | EFO:0000434 | TRUE | FALSE |
| env_broad_scale | biome | ENVO:00000428 | FALSE | TRUE |
| genotype | genotype | SIO:001079 | TRUE | FALSE |
| geo_loc_name | geographic location | data:3720 | TRUE | TRUE |
| growth_protocol | growth protocol | EFO:0003789 | TRUE | FALSE |
| height_or_length | height/length | SIO:000040 height, SIO:000041 length | TRUE | FALSE |
| host | host | SIO:010415 | FALSE | TRUE |
| host_tissue_sampled | FALSE | TRUE | ||
| identified_by | FALSE | TRUE | ||
| isolate | TRUE | TRUE | ||
| isolation_source | Isolation source | data:3721 | TRUE | TRUE |
| lat_lon | Longitude, latitude | SIO:000318, SIO:000319 | TRUE | TRUE |
| organism | organism | OBI:0100026 | TRUE | TRUE |
| phenotype | phenotype | SIO:010056 | TRUE | FALSE |
| population | population | SIO:001061 | TRUE | FALSE |
| sample_name | name | schema:name | TRUE | TRUE |
| sample_title | title | SIO:000185 | TRUE | TRUE |
| sample_type | rdfs:type | TRUE | FALSE | |
| sex | biological sex | SIO:010029 | TRUE | TRUE |
| specimen_voucher | TRUE | TRUE | ||
| temp | temperature | EFO:0001702 | TRUE | TRUE |
| tissue | tissue | SIO:010002 | TRUE | TRUE |
| treatment | treatment | EFO:0000727 | TRUE | FALSE |
Where no ontology terms were found, fields are left blank.
Mapping NCBI’s Taxon and PubMed content
NCBITaxon and PubMed records, which contain relevant metadata associated with genome assemblies, are already imported by the Tripal core importers. These data are imported into the Chado organism and pub tables, respectively. In both cases, the mapping is intuitive. We document the mapping logic in Supplementary Tables S1–3.
Tripal EUtils
We have written a Tripal extension module that imports NCBI metadata into Chado using the NCBI Entrez Programming Utilities (E-utilities) (7,17). The module provides a web form for Tripal site administrators to query NCBI’s E-utilities. The administrator supplies an NCBI accession or unique ID for any of the supported data types previously discussed and clicks the preview button. The module queries NCBI’s E-utilities, parses the returned record and provides a preview display to the administrator. Additionally, some NCBI records refer to records in other resources. For example, a BioProject might link to the accession for an Assembly and BioSample. NCBITaxon and PubMed resource accessions may also be linked. From the preview display page, the administrator can then specify if he or she would like to insert the record—the linked records—and then submit the job. Tripal EUtils inserts the retrieved NCBI record, and any linked records, into the Chado database as specified by the above mappings.
The Tripal EUtils module relies on NCBI’s E-utilities service for metadata retrieval (7). The module is available at https://github.com/NAL-i5K/tripal_eutils under a GPL-3 license. E-utilities’ support for individual NCBI databases or resources was implemented using instructions provided in the NCBI E-utilities developer’s guide (https://www.ncbi.nlm.nih.gov/books/NBK25500/).
To test the Tripal EUtils functionality, 70 NCBI BioProjects under the i5k Arthropod Genome Pilot Project (PRJNA163973) and their linked Taxons, Assemblies, Publications and BioSamples were used. The locust BioProject (uid, 91111; accession, PRJNA185471) and its associated records are presented in screenshots that follow as examples.
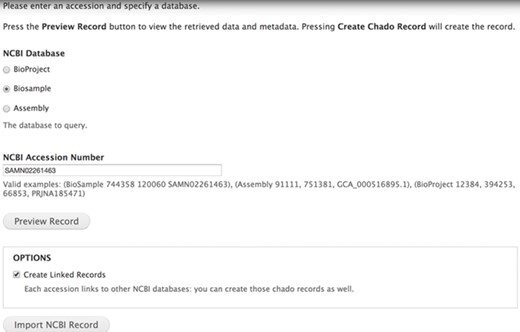
The Tripal EUtils accession importer. Administrators select a database and provide a numeric uid or mixed accession. Clicking the Preview button lists all the fields, properties and additional links found in the record. Each record links back to NCBI. Administrators can choose to only import the single record or to also insert records linked to that primary record. In this example, BioSample accession SAMN02261463 will be inserted into Chado.
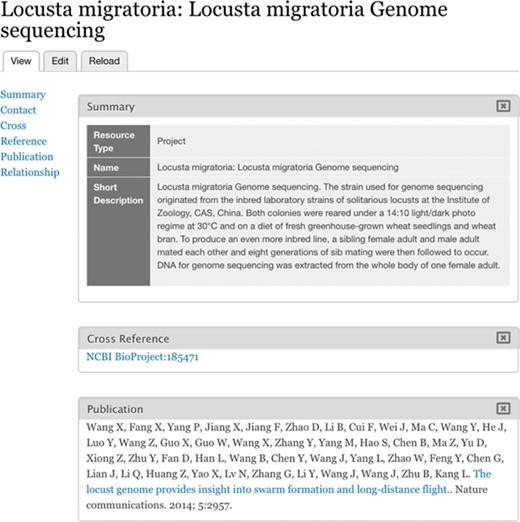
Imported BioProject. BioProject 185471 is stored as a Chado Project record.
The Tripal EUtils module is meant to facilitate easy adoption of these proposed mappings. Thus, all Tripal sites can easily adopt the metadata standards we propose here. Moreover, Tripal site developers with PHP coding experience, who wish to add mappings for other NCBI databases can do so by (i) extending a new EUtil XML parser class, which reads the databases’ returned XML; and (ii) extending a new repository and formatter class, which describes the metadata to the user and inserts into Chado.
Interface
The Tripal EUtils module provides a form for administrators to preview and insert individual accessions for the Assembly, BioProject and BioSample resources. After providing the accession, the form populates a preview of the record to be created, summarizing the record and its associated properties (Figure 1). Once approved, the metadata are inserted into the Project (Figure 2), Biomaterials (Figure 3), Analysis (Figure 4), Taxon (Figure S1) and Pub (Figure S2) tables.
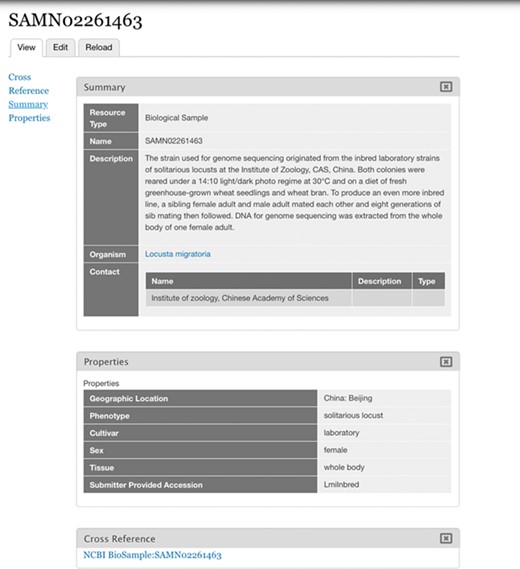
Imported BioSample. BioSample SAMN02261463 represented as a Chado Biomaterial/Tripal Sample bundle as imported by the Tripal EUtils module. The properties imported are for geographic location, phenotype, cultivar, sex, tissue and submitter provided accession.
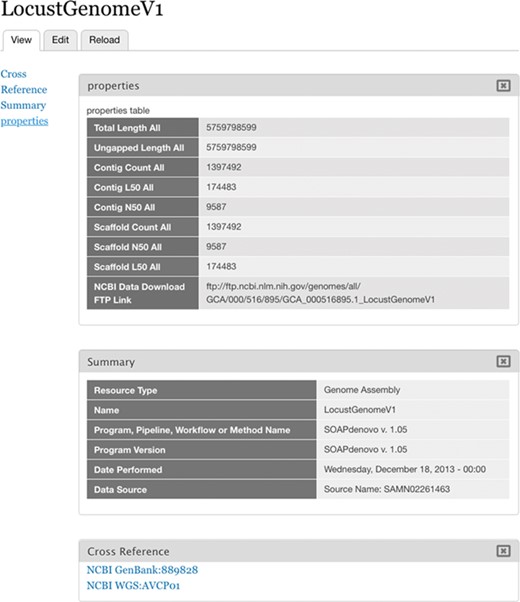
Imported Assembly. Assembly WGS AVCP01 as represented in Chado.
Documentation
The EUtils module is fully documented and available at https://tripal-eutils.readthedocs.io/en/latest. Documentation is built using Sphinx and hosted with ReadTheDocs (https://readthedocs.org/). The User’s Guide provides installation, setup and usage instructions. Each supported NCBI database has a guide describing the metadata mappings as detailed in Tables 1–4. The Developer’s Guide includes all information needed for adding support for new NCBI databases. The Developer’s Guide is available at https://tripal-eutils.readthedocs.io/en/latest/code_doc/eutils.html.
Modifying metadata in Chado
The Tripal EUtils module does not allow the admin user to modify the metadata from NCBI during import. It is possible for the admin user to edit the instantiated record after import; however, we caution against doing so without also updating the metadata at the corresponding NCBI resource. Conflicting metadata records at different databases are likely to sow confusion for end users.
Future direction
Proposed additions to Chado
We believe that Chado can offer a model for storing records hosted by NCBI. Unfortunately, we have found that some concepts may require modifications to the Chado table definitions. An NCBI Assembly or Genome record, for example, may be the result of a series of separate programs or analyses that a researcher conducted; the results of which were combined to generate the final genome assembly. However, as stated previously, an analysis record in Chado should explicitly be the execution of a single computational program. This implies we should create a record in the analysis table for each individual program run in the pipeline.
There are several possibilities to amend Chado to readily accommodate metadata describing such data. First, an assembly, for example, is a set of steps executed in order. While not automated, it constitutes the output of a scientific ‘workflow’. The analysis table definition could therefore be broadened to encompass a scientific workflow. Second, Chado could introduce a new table designed to describe workflows. The table would be designed for storing versioned, linked analytical pipelines, their parameters, input data and output data. Such a table could be cross-compatible with existing experimental data models such as ISA-commons (18) or COPO (https://copo-project.org/).
From ingestion to export
This paper has focused on providing a convenient means for importing metadata from NCBI into a Chado database hosted by a Tripal site. Many of the features Tripal offers for searching, organizing and ontologizing datasets are well suited for exporting data and metadata back into NCBI as well. A Tripal community database has the means to heavily structure the end user’s data submission and annotation process, package it and submit it to NCBI. Future Tripal modules for NCBI submission may facilitate this. For example, a community might agree to require specialized, field-specific property types for their BioSample submissions. The end result would be a streamlined user experience but with better, community-specialized adherence to data and metadata best practices.
Module sustainability
The current version of Tripal, version 3.1, is based on Drupal version 7, which will not be supported after November 2021. We anticipate that Tripal core development will continue toward supporting Drupal version 8. Tripal 3.1 was developed to be forward compatible with Drupal 8; we therefore expect that updating the Tripal EUtils module to Drupal 8 will be straightforward.
Conclusions
We have presented metadata maps to better reconcile genome assembly metadata models between NCBI and Chado. The resulting Tripal EUtils module provides a means for Tripal databases to store NCBI-derived metadata in a consistent manner across databases. Ultimately, this should facilitate (i) better interoperability among Tripal databases, (ii) easier data and metadata retrieval by users across Tripal databases due to increased consistency and (iii) reduced development time for other Tripal databases with similar use cases. We hope that this mapping lays the groundwork for a standard way to store genome assembly metadata across Tripal databases, to be adopted by the Chado community.
Acknowledgements
We thank Ethalinda Cannon, Gerard Lazo and three anonymous reviewers for the thoughtful reviews of this manuscript.
Funding
USs Department of Agriculture’s National Agricultural Library; National Science Foundation (1444573 to M.S.).

{kind=link}
{kind=link}
{kind=link}
{kind=link}