





Abstract
DNA metabarcoding is a widespread approach for the molecular identification of organisms. While the associated wet-lab and data processing procedures are well established and highly efficient, the reference databases for taxonomic assignment can be implemented to improve the accuracy of identifications. Insects are among the organisms for which DNA-based identification is most commonly used; yet, a DNA-metabarcoding reference database specifically curated for their species identification using software requiring local databases is lacking. Here, we present COins, a database of 5’ region cytochrome c oxidase subunit I sequences (COI-5P) of insects that includes over 532 000 representative sequences of >106 000 species specifically formatted for the QIIME2 software platform. Through a combination of automated and manually curated steps, we developed this database starting from all COI sequences available in the Barcode of Life Data System for insects, focusing on sequences that comply with several standards, including a species-level identification. COins was validated on previously published DNA-metabarcoding sequences data (bulk samples from Malaise traps) and its efficiency compared with other publicly available reference databases (not specific for insects). COins can allow an increase of up to 30% of species-level identifications and thus can represent a valuable resource for the taxonomic assignment of insects’ DNA-metabarcoding data, especially when species-level identification is needed https://doi.org/10.6084/m9.figshare.19130465.v1.
Introduction
DNA metabarcoding is a popular method in molecular taxonomy widely used for organisms’ identification starting from short DNA sequences of one or a few genes (1, 2). This method has wide applicability in many different fields in which the identification of living organisms is required (3–6). DNA-based identification methods are more useful on organisms for which identification using other approaches is problematic, requires vast expertise or takes a long time. Due to their species richness and ubiquity and to the high level of specialization required for their morphological identification, insects represent one of the groups for which DNA-based identification is commonly used (7–10).
Currently, DNA-metabarcoding wet-lab protocols and raw data analysis pipelines are well established, allowing researchers to obtain high-quality results both from insect environmental DNA and insect community DNA samples (11). Nevertheless, the choices of the DNA marker(s) and of the reference database for sequences’ taxonomic assignment are two key aspects that can affect the accuracy of identifications.
Depending on the aim of the study, different DNA markers are deemed as appropriate for insect molecular identification (12, 13). Some of them have a wide taxa coverage but a low taxonomic resolution, e.g. 16S ribosomal RNA (rRNA) and 18S rRNA (13, 14), while others permit more specific identifications, e.g. cytochrome oxidase subunit I (COI (14)). The choice of the marker is usually driven by the amount of data available as reference, in particular when prior knowledge on the sampled insect taxa is lacking (as in the case of biodiversity surveys; insectivorous animals’ diets characterization (15, 16)). In this case, COI is the best choice thanks to the high number of publicly available sequence data stored within online repositories (major ones being Barcode of Life Data (BOLD) System and GenBank (17, 18)), especially for the 5′ end (COI-5P), the region that can be amplified using universal DNA-barcoding primers, such as LCO1490/HCO2198 (19). In recent years, a consistent number of DNA-metabarcoding primers targeting this region have been developed (20, 21) and demonstrated to work effectively on different insect taxa (21).
Regarding reference databases of COI sequences, some types of software directly connecting to BOLD system databases for taxonomic assignments of operational taxonomic units (OTUs)/amplicon sequence variants (ASVs) are available (22, 23), but other tools (e.g. QIIME (24), Ribosomal Database Project (RDP) classifier (25) BLAST+ (26)) need local reference databases. For the latter, some publicly available ready-to-use databases have been developed (e.g. MIDORI (27)). Nevertheless, in some cases, the use of self-developed reference databases can increase taxa identification accuracy. Generating a customized reference database can be a challenging process, and until recently, it has been possible only using self-developed pipelines. Recently, Robeson and colleagues (28) released RESCRIPT, a largely automated tool for creating metabarcoding reference databases starting from online repositories of public sequence data. Exploiting the large amount of publicly available data for developing DNA-metabarcoding reference databases for insects’ identification can be convenient, considering that the identification success and accuracy using DNA metabarcoding is strongly dependent on the completeness of the reference database in terms of taxa representative sequences (29). Indeed, such kinds of references for insects can be developed de novo only by combining the forces of multiple researchers having different taxonomic competence, due to the high taxa richness and intra-taxon diversity (in terms of COI variability) characterizing this group. Although the large amount of COI sequence data stored in online repositories may sound like a fundamental asset for creating a good reference database, dealing with data developed by other people and generated in the context of different studies can be a double-edged sword. In fact, sequences of undesired origin [nuclear mitochondrial pseudogenes (numts) and contaminants] or related to the wrong taxonomy are commonly released (11), and identifying them among a huge amount of data can be challenging. The presence of erroneous sequences in a reference database can lead to the misidentification of taxa. However, fully automatizing their filtering within a bioinformatic pipeline is unlikely to succeed; as a result, a manual curation step is always fundamental (28).
Here, we present COins, a curated reference database for insect molecular identification that can be used with software requiring a local reference database. This novel database leverages the COI sequence of the 5′ region published on BOLD and has been developed using a combination of automated and manual curation steps. The goal was to develop a tool allowing more accurate and specific identification to be obtained than by using the resources currently available for the molecular identification of insect taxa.
Materials and methods
Data mining and database curation
All DNA sequences of insects publicly available on the BOLD COI database were downloaded (search query ‘Insecta’) on 18 September 2020 along with the information on the specimens from which they were generated. All subsequent steps were performed through ad hoc bash and R software (R Core Team, 2019) scripts unless otherwise specified. From the mined dataset, COI-5P and full gene sequences were selected, and any sequences that lacked the relevant taxonomic information on the specimens they were developed from (i.e. order, family, genus and species of belonging) were removed. Then, multi-FASTA format files were generated for each insect order included in the dataset, and the related taxonomy was reported as sequences ids (insect orders sub-datasets, Step 1; Figure 1). In order to avoid the presence within the datasets of sequences annotated as insect but actually belonging to other organisms, the sequences were compared with selected Homo sapiens, Wolbachia and Rickettsia sequences using BLAST+ (30), and the entries matching non-insect sequences with an e-value < 1e−20 were removed (Step 2; Figure 1). Additionally, sequences >150 amino acids were identified using Transeq from the EMBOSS software (31) and removed (Step 2; Figure 1). As a further step, the sequences associated with an invalid species-level taxonomy were identified and removed using two methods: (i) an ad hoc script looking for key terms included in species names, i.e. ‘sp.’, ‘cf.’, ‘cfr.’, ‘group’, ‘nr.’ and numbers and (ii) a manual filtering for detecting further invalid names non-identifiable through key terms (e.g. collection localities, collectors or species author names instead of species name, alphabetic codes replacing species name and many others) (Step 2; Figure 1). To further verify sequences homology, each dataset was aligned at the codon level using MAFFT software (32), and the identity of all sequences introducing gaps in the alignments was verified using the BLASTn tool (33) and, in case of incongruence, removed (Step 3; Figure 1). The resulting datasets were then trimmed to keep only the COI gene ‘Folmer region’ (19); all sequences having a length of ≤420 bp were then discarded using the R library spider (34) (Step 4; Figure 1). In the subsequent step, one representative sequence for each haplotype of the species included in the datasets was selected using the R package haplotypes (https://biolsystematics.wordpress.com/r/) (Step 5; Figure 1). All insect orders’ sub-datasets were then combined into a single multi-FASTA file in which BOLD process ids were used as sequences’ identifiers, and sequences’ associated taxonomy was stored in a separate file (Step 6; Figure 1). Within this file, five Rickettsiales sequences amplified from insects using the Folmer primers pair (19) were also included. Due to these sequences’ similarity to insects’ COI-5P, including them in the database could allow the avoidance of misassignment. Finally, the database was formatted for QIIME2 (.qza files available at https://doi.org/10.6084/m9.figshare.19130465.v1). Any further updated version of the database will be published at the same link. COins will be updated whenever a meaningful number of COI insect sequences will be published on the BOLD system.
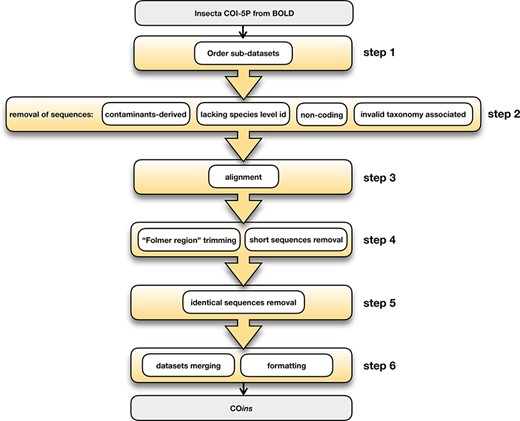
COins database development steps.
Reference database efficiency test
DNA-metabarcoding raw data (obtained from 54 bulk samples collected with Malaise traps) developed in Kirse et al. (35), using mlCOIintF (5′–ACA CTC TTT CCC TAC ACG ACG CTC TTC CGA TCT GGW ACW GGW TGA ACW GTW TAY CCY CC–3′) and dgHCO2198 (5′–GTG ACT GGA GTT CAG ACG TGT GCT CTT CCG ATC TTA AAC TTC AGG GTG ACC AAA RAA YCA–3′) primers pair, were obtained from the Sequence Read Archive (SRA) archive (project accession number PRJNA68109) and used to test the efficiency of the developed database. The bioinformatic analyses were performed using the QIIME2 platform (24). Raw sequences were denoised with the DADA2 algorithm (36) to remove errors and obtain the actual biological sequences (ASVs).
The ASV taxonomic assignment was then performed using two approaches: (i) BLAST+ local alignment between query and reference reads (sequence identity = 97%, minimum consensus among top hits = 80% (26)) and (ii) the naïve Bayes taxonomic classifier trained on the reference database using the fit-classifier sklearn method (confidence = 0.97 (37, 38)). Three different databases were used as reference: (i) the database developed in this study, hereafter named COins; (ii) MIDORI CO1 unique version 245 (Leray et al., in preparation; http://www.reference-midori.info/download.php#) and (iii) a reference database of COI sequences created using RESCRIPT software starting from animals’ COI sequences registered in BOLD (retrieving date July–August 2020), hereafter named ResBO (database available at https://osf.io/d4jra/).
Results
The database
A total of 5 065 234 insect COI sequences were mined from BOLD. After filtering (up to Step 4; Figure 1), 3 745 421 sequences were lost (mainly due to the removal of sequences lacking species-level identification). At the end of Step 6 (Figure 1), the database was composed of 532 617 unique sequences, belonging to >106 000 species of 27 different insect orders. The most represented order within COins is Lepidoptera, followed by Diptera and Coleoptera (Table 1). Only a few sequences of Zoraptera and Notoptera are present (Table 1).
Number of unique sequences for each insect order included in the database
| Order | Number of sequences |
|---|---|
| Archaeognatha | 79 |
| Blattodea | 1558 |
| Coleoptera | 65 684 |
| Dermaptera | 140 |
| Diptera | 122 306 |
| Embioptera | 69 |
| Ephemeroptera | 7150 |
| Hemiptera | 28 494 |
| Hymenoptera | 58 124 |
| Lepidoptera | 209 290 |
| Mantodea | 378 |
| Mecoptera | 304 |
| Megaloptera | 281 |
| Neuroptera | 1821 |
| Notoptera | 3 |
| Odonata | 5142 |
| Orthoptera | 7369 |
| Phasmatodea | 172 |
| Plecoptera | 4733 |
| Psocodea | 1800 |
| Raphidioptera | 41 |
| Siphonaptera | 473 |
| Strepsiptera | 56 |
| Thysanoptera | 1778 |
| Trichoptera | 15 321 |
| Zoraptera | 2 |
| Zygentoma | 49 |
| Order | Number of sequences |
|---|---|
| Archaeognatha | 79 |
| Blattodea | 1558 |
| Coleoptera | 65 684 |
| Dermaptera | 140 |
| Diptera | 122 306 |
| Embioptera | 69 |
| Ephemeroptera | 7150 |
| Hemiptera | 28 494 |
| Hymenoptera | 58 124 |
| Lepidoptera | 209 290 |
| Mantodea | 378 |
| Mecoptera | 304 |
| Megaloptera | 281 |
| Neuroptera | 1821 |
| Notoptera | 3 |
| Odonata | 5142 |
| Orthoptera | 7369 |
| Phasmatodea | 172 |
| Plecoptera | 4733 |
| Psocodea | 1800 |
| Raphidioptera | 41 |
| Siphonaptera | 473 |
| Strepsiptera | 56 |
| Thysanoptera | 1778 |
| Trichoptera | 15 321 |
| Zoraptera | 2 |
| Zygentoma | 49 |
Number of unique sequences for each insect order included in the database
| Order | Number of sequences |
|---|---|
| Archaeognatha | 79 |
| Blattodea | 1558 |
| Coleoptera | 65 684 |
| Dermaptera | 140 |
| Diptera | 122 306 |
| Embioptera | 69 |
| Ephemeroptera | 7150 |
| Hemiptera | 28 494 |
| Hymenoptera | 58 124 |
| Lepidoptera | 209 290 |
| Mantodea | 378 |
| Mecoptera | 304 |
| Megaloptera | 281 |
| Neuroptera | 1821 |
| Notoptera | 3 |
| Odonata | 5142 |
| Orthoptera | 7369 |
| Phasmatodea | 172 |
| Plecoptera | 4733 |
| Psocodea | 1800 |
| Raphidioptera | 41 |
| Siphonaptera | 473 |
| Strepsiptera | 56 |
| Thysanoptera | 1778 |
| Trichoptera | 15 321 |
| Zoraptera | 2 |
| Zygentoma | 49 |
| Order | Number of sequences |
|---|---|
| Archaeognatha | 79 |
| Blattodea | 1558 |
| Coleoptera | 65 684 |
| Dermaptera | 140 |
| Diptera | 122 306 |
| Embioptera | 69 |
| Ephemeroptera | 7150 |
| Hemiptera | 28 494 |
| Hymenoptera | 58 124 |
| Lepidoptera | 209 290 |
| Mantodea | 378 |
| Mecoptera | 304 |
| Megaloptera | 281 |
| Neuroptera | 1821 |
| Notoptera | 3 |
| Odonata | 5142 |
| Orthoptera | 7369 |
| Phasmatodea | 172 |
| Plecoptera | 4733 |
| Psocodea | 1800 |
| Raphidioptera | 41 |
| Siphonaptera | 473 |
| Strepsiptera | 56 |
| Thysanoptera | 1778 |
| Trichoptera | 15 321 |
| Zoraptera | 2 |
| Zygentoma | 49 |
Two metadata files associated with COins are available. The first one comprises the information on the identification procedure of the voucher specimens from which COI sequences included in the database were generated. The same information is reported also for all identical sequences within haplotypes that were removed in Step 5 of the database curation (Figure 1). The second file reports the information on identical sequences belonging to different species present within the database. These files can be consulted when any specific molecular identification obtained using COins is doubtful (available at https://doi.org/10.6084/m9.figshare.19130465.v1).
Database efficiency test
The 54 DNA-metabarcoding samples (32) used to test the database efficiency, included a total of 27 348 365 raw reads (mean per sample = 506,451.2 reads), after denoising and filtering 8312 ASVs were obtained. The two algorithms adopted in this study (BLAST+-based and fit-classifier sklearn) demonstrated a good congruence in the taxonomic assignments of the ASVs detected, with COins sharing the highest number of ASVs’ unique identifications between algorithms than the other databases, i.e. 80.6% in comparison to 73.6% for MIDORI and 67.8% for ResBO (Figure 2).
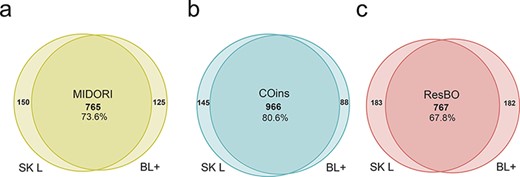
Number of ASVs identified by the two taxonomic assignment algorithms adopted in this study, i.e. the machine learning-based algorithm fit-classifier sklearn (SK L) and the BLAST+ (BL+) algorithm, using each database: (a) MIDORI database, (b) COins database and (c) ResBO database. Numbers of common identifications between the two algorithms are also expressed in percentages.
The taxonomic assignments of these ASVs using as reference ResBO resulted in 2381 (using BLAST+ algorithm) and 2870 (fit-classifier sklearn algorithm) ASVs assigned to the Insecta class. COins identified 2368 (BLAST+) and 8026 (fit-classifier sklearn) Insecta ASVs, while MIDORI identified 1876 (BLAST+) and 3273 (fit-classifier sklearn) ASVs. Among them, order-level assignments were obtained for 2374 (BLAST+) and 2008 (fit-classifier sklearn) ASVs adopting ResBO as reference; 2367 (BLAST+) and 2611 (fit-classifier sklearn) ASVs using COins and 1864 (BLAST+) and 2219 (fit-classifier sklearn) ASVs using MIDORI (Figure 3). Regarding species-level assignments, the following results were obtained: ResBO identified 1530 (BLAST+) and 1608 (fit-classifier sklearn) ASVs to species; COins 2117 (BLAST+) and 2243 (fit-classifier sklearn) ASVs to species and MIDORI 1594 (BLAST+) and 1584 ASVs (fit-classifier sklearn) to species (Figure 3).
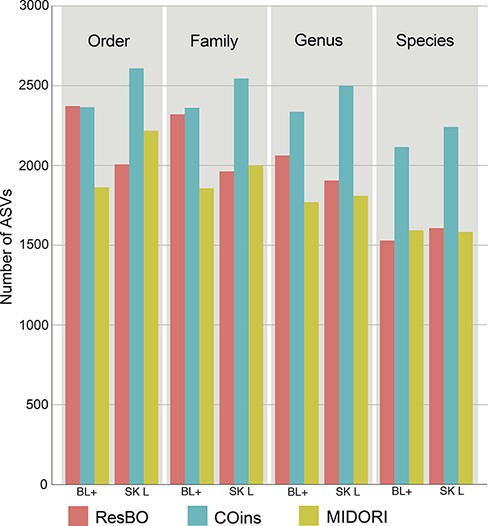
Number of ASVs assigned to the different taxonomic levels (from order to species) when using ResBO, COins and MIDORI as reference. Numbers of assignments obtained using the BLAST+ (BL+) and fit-classifier sklearn (SK L) algorithms are specified too.
Among the species-level identified ASVs using BLAST+-based algorithm, 825 different species were recognized by MIDORI: 27 of them were shared with ResBO, which identified 887 species (Figure 4a). The highest number of species was found using COins, i.e. 1051, 184 of them in common with ResBO (Figure 4a). Using the BLAST+-based algorithm, 41.4% of the species were identically identified by the three reference databases (Figure 4a). A similar situation was observed when fit-classifier sklearn algorithm was applied, in fact 836 different species were identified by MIDORI (Figure 4b), 29 of them were shared with ResBO, which identified 866 species, and COins detected 1108, 202 in common with the last database (Figure 4b). Using this algorithm, the percentage of common species recognized by the three databases was 40.1% (Figure 4b).
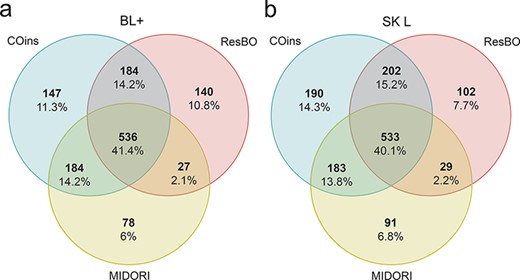
Number of species identified using each database MIDORI, COins and ResBO. (a) Number of species identified adopting the BLAST+ algorithm (BL+). (b) Number of species identified adopting fit-classifier sklearn algorithm (SK L). All values are also reported as percentages.
COins identified some ASVs as belonging to Rickettsiales (<20), these ASVs were assigned to Insecta, Arthropoda or remained unassigned when using the other reference databases.
Discussion
In this study, a reference database of COI sequences (5′ region) for insects’ taxonomic assignment using DNA metabarcoding was developed, starting from the data available on BOLD. These data were filtered according to several criteria in order to remove sequences, which might be potential sources of error during taxonomic assignments of the ASVs. Different motivations for sequence removal—along with their implications—are discussed below.
Sequences associated with incorrect or invalid taxonomy. The most common situation was the presence of sequences annotated as insect but instead derived from other organisms, in particular Homo sapiens and also the most common bacterial endosymbionts of insects (e.g. Wolbachia and Rickettsia). The latter is an already well-known problem related to online reference databases (39). Filtering COI sequences separately as sub-datasets for each insect order allowed us to detect further inconsistencies between sequences’ variability and their associated taxonomy. In particular, during the alignment step, some sequences showing low overall homology with the others in the same sub-dataset were found to be related to misidentifications at the order level. Within this study framework, the official validity of all sequences-associated taxonomic names was intentionally not investigated, because of ongoing debates on the taxonomic status of some insect taxa. As a matter of fact, the increasingly common use of molecular taxonomy has introduced a bias in insect taxonomy: frequently, new species are recognized based on molecular information (e.g. through molecular species delimitation or in the context of DNA-barcoding studies) and named, but never, or only much later, formally described. These species names are not considered valid according with the International Code of Zoological Nomenclature (40) until the formal description of the species is published, but online databases include the reference sequences which allow their identification under the new species name. Nonetheless, the filters applied to the sequences, the manual filter in particular, allowed the detection and discarding of many invalid species names unrelated to the above-mentioned situations and possibly linked with the absence of species-level morphological identification (e.g. genera names followed by numeric or alphabetic codes, but also geographical names or person names replacing specific epithets). In case of doubt, the scientific works within which the sequences were developed were consulted.
Non-coding sequences were possibly derived from the amplification of numts (41), from sequencing errors, or from the lack of proper editing of electropherograms before data publication. This issue was particularly evident in the database alignment step, where many sequences were discarded since they introduced one or two bases’ gaps in the alignment.
Sequences not associated with species-level taxonomy within a reference database, especially if identified at the highest taxonomic ranks, appear to reduce the accuracy of the molecular identification, hindering the reaching of identifications at lower taxonomic levels. This scenario is also a likely explanation for some of the results achieved in the present study, i.e. the cases in which COins assigned the ASV at the species level, while ResBO assigned the same ASVs to a higher taxonomic level, despite the two databases include the same species-level identified reference sequences. At the same time, excluding from a reference database, the sequences not identified at the species level could potentially increase the number of missing identifications, especially when those sequences belong to the only representative of a specific taxon within the database.
Some of the sequences discarded from the database are clearly related to errors, and they could be the results of the lack of care of some BOLD users, as indeed is also a common situation in the case of other databases. The BOLD team routinely perform data curation, in particular checking discordant Barcode Index Number and suppressing potential erroneous sequences from the online database (42). As in the case of this study, the curation is performed manually. It is a time-consuming process done periodically, thus leaving some erroneous sequences in place for a while. This is why using publicly available data for developing DNA-metabarcoding reference databases for local use should always require a manual curation step (28).
The efficiency test on COins showed how this database has an identification efficiency comparable to that of the other databases (MIDORI and ResBO) at the highest taxonomic ranks (e.g. order and family), but it allows the assignment of a considerably higher number of ASVs to the species and genus levels, with a notable increase between 25% and 30% of species-level identifications.
The performed analyses also allowed observation to be made on the effect of using different assignment algorithms. The machine learning-based algorithm (fit-classifier sklearn) was found to assign a higher number of ASVs at any taxonomic level, compared with the BLAST+ algorithm (Figure 3). An evident bias of the use of the fit-classifier sklearn algorithm in association with COins is that almost all the ASVs detected in the samples analysed were assigned to Insecta (8026 ASVs out of 8312) even if some of them likely belong to other classes (e.g. sequences that MIDORI and ResBO assigned to Collembola or Arachnida). This is related to the underlying principle of machine learning-based algorithms, which assumes that all existing taxa are included in the reference used for the assignment (37, 38). Yet, this drawback is only associated with higher level taxonomic assignments and does not affect the accuracy of low-level ones. As a matter of fact, COins was the database for which the highest congruence between identification achieved through the two algorithms used in this study was achieved (Figure 2).
The results obtained using COins highlight the importance of manual curation during the development of reference databases for local use. The effort required is however undeniable. Unfortunately, fully automated filters that make sequences downloaded from public resources readily usable for metabarcoding taxonomic assignment are not yet available. In the meantime, it is necessary, albeit expensive and time-consuming, especially in terms of updating, to make high-quality data available for those metabarcoding software platforms that use local reference databases. Moreover, the direct interaction between software such as QIIME2 with the online BOLD COI database for metazoan ASV/OTU taxonomic assignment is also advisable.
Funding
The authors acknowledge the support of the Article Processing Charge (APC) central fund of the University of Milan (Italy) and the Department of Agricultural and Environmental Sciences of the University of Milan (Italy) which provided the postdoc fellowship of the first author (years 2020–2022).
Conflict of interest
None declared.

{kind=link}
{kind=link}
{kind=link}
{kind=link}