





Abstract
Tracking scientific research publications on the evaluation, utility and implementation of genomic applications is critical for the translation of basic research to impact clinical and population health. In this work, we utilize state-of-the-art machine learning approaches to identify translational research in genomics beyond bench to bedside from the biomedical literature. We apply the convolutional neural networks (CNNs) and support vector machines (SVMs) to the bench/bedside article classification on the weekly manual annotation data of the Public Health Genomics Knowledge Base database. Both classifiers employ salient features to determine the probability of curation-eligible publications, which can effectively reduce the workload of manual triage and curation process. We applied the CNNs and SVMs to an independent test set (n = 400), and the models achieved the F-measure of 0.80 and 0.74, respectively. We further tested the CNNs, which perform better results, on the routine annotation pipeline for 2 weeks and significantly reduced the effort and retrieved more appropriate research articles. Our approaches provide direct insight into the automated curation of genomic translational research beyond bench to bedside. The machine learning classifiers are found to be helpful for annotators to enhance the efficiency of manual curation.
Introduction
Advances in human genomic research promise a new era of precision medicine and personalized healthcare. Beyond basic discoveries and development of genomic tests and related intervention, the success of genomic medicine will increasingly depend on translational research beyond bench to bedside, including studies of clinical validity and utility, and the study of dissemination and implementation of evidence-based genomic applications (1). In the rapidly developing field of translational genomic medicine, finding and curating related research is highly critical for improving health care and population health (2, 3). However, this curation task requires knowledge of multiple disciplines, such as clinical trials, epidemiology, behavioral and social science, health services research, implementation science and economics. The workflow of collecting and integrating data is both time-consuming and costly through manual curation. As a result, the rapid growth of biomedical literature creates barriers to the transfer of knowledge between basic discoveries and public health impact.
One classification of translational research continuum involves four phases beyond an initial discovery. T1 research involves the development of candidate applications such as tests, treatments and other interventions (classical bench to bedside); T2 research evaluates the clinical utility of candidate applications leading to evidence-based recommendations for practice; T3 research integrates evidence-based recommendations into clinical practice (implementation science); and T4 research assesses the outcomes and population impact of genomics in the real world (2). Note that `beyond bench-to-bedside’ phases (T2–T4) include the evaluation, utility and implementation of evidence-based applications into clinical practice and evaluation of the impact these applications have on population health. However, it is reported that <2% of the published literature on human genomics is within T2–T4 (1).
To develop a baseline for progress in translation of genomic medicine into practice, the Office of Public Health Genomics (OPHG) of the Centers for Disease Control and Prevention in collaboration with the National Cancer Institute and the National Heart, Lung and Blood Institute have been regularly curating the genomic translational literature since 2012, which is housed in the Public Health Genomics Knowledge Base (PHGKB) (4).
With the increasing number of genomic research articles from bench to bedside and beyond, the workload for manual curation of articles relevant to the latter stages of translational research has also increased exponentially. In this work, we aim to assist manual curation with automated text mining methods, as successfully demonstrated by few recent studies (5–7). Specifically, we employ previously annotated data sets by PHGKB and develop machine learning approaches to automatically classify the articles belonging to T2–T4 phases.
Supervised machine learning methods such as support vector machine (SVM) have been shown to be effective for document classification (8). Surkis et al. (9) categorized translational publications from T0 to T4 by applying machine learning-based text classifiers, and the SVM achieved the best performance among the comparison methods. However, there are some limitations in such a method. For instance, the `bag-of-words’ feature in their method did not fully capture semantic or syntactic information. Second, their method made use of the medical subject headings (MeSH) indexing terms as a feature, but MeSH terms, generally speaking, are not immediately available upon article publication (10). Nonetheless, we include the SVM as a comparison algorithm given its superior performance in the previous study.
More recently, deep neural network-based approaches have shown improved results in many natural language processing (NLP) tasks including text classification (11). Neural network-based approaches such as the convolutional neural network (CNN) have been applied to assist document triage of kinome curation, genomic variation and protein–protein interactions (12–14). Lee et al. (13) employed the CNN to identify publications that are relevant for variant curation. They demonstrated that the deep learning-based classifier outperforms traditional machine learning classifiers without feature engineering. Luo et al. (14) used the ensemble of the neural network models to achieve state-of-the-art performance on the document triage task of BioCreative VI Track 4: mining protein interactions and mutations for precision medicine (15). Hence, in this work, we propose to capture the characteristics of beyond bench-to-bedside phase articles using a CNN-based approach (16). Through evaluation, we find that our proposed method is highly accurate and has the potential to greatly improve the current workflow of curating beyond bench-to-bedside articles in genomic translational research.
Materials and methods
Data sets
The training and test data sets for the translational phases classification (TPC) task were provided by OPHG. The data sets were previously collected, reviewed and annotated by The Centers for Disease Control and Prevention (CDC) curators according to the schema defined in (2). In the TPC task, the articles are classified into two separate translational phases: the initial bench-to-bedside phases (T1) and the beyond bench-to-bedside phases (T2–T4). In total, the training data set consists of 2286 articles, including 1379 articles in T1 and 907 articles in T2–T4 (published from 23 August 2016 to 14 October 2016). The test set consists of 400 articles, including 241 articles in T1 and 159 articles in T2–T4 (published after October 2016).
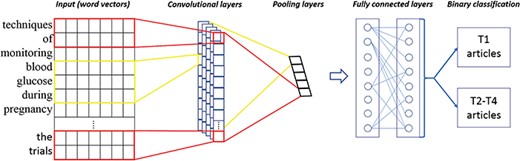
The CNNs for the TPC task.
Machine learning algorithms
We formulate the TPC task as a binary document classification task and experiment with two machine learning algorithms. SVMs are supervised learning models based on a statistical learning theory (17), and it has been used to many text classification problems with a state-of-the-art performance. The SVMs usually involve training samples with many features and labeled classes (18). Next, the SVMs find a hyperplane that divides the sample into different groups and produce an inferred function to test the characteristics of the samples. After model training, the classifier can efficiently predict the correct class labels for unknown instances. In this work, linear SVMs are used as a baseline method and we train them using the article title and abstract with the bag-of-words model. In addition, we have also tested features such as the journal title, but none was able to result in an improved performance overall. We use the 5-fold cross-validation results on the training set to choose the best parameters (C = 1000) for our SVM classifier, e.g. we search for the best value for C (cost) value to avoid overfitting by balancing the performances on training and test data.
CNNs are one kind of artificial neural networks. Figure 1 gives an illustration of the architecture of our CNN-based classifier, derived from (19). The main characteristics of CNNs are convolutional layers, pooling layers and fully connected layers. The former detects patterns in multiple subregions and extracts salient features by filter weights, which generate a large amount of feature maps. After the convolutional layers, the pooling layers down-sample the feature maps and control overfitting by reducing the number of parameters. Next, the fully connected layers are responsible for transforming and voting important features from convolutional and pooling layers. The fully connected layers consist of flatten, hidden and softmax output layers, and we can obtain predicted class probabilities at the last stage. Note that the fully connected layers often involve with a large amount of computation, such as adjusting weights of networks and connecting strength between neurons (backpropagation). In recent years, CNNs have been shown to be effective in many NLP tasks, such as sentence modeling and classification (20, 21). In this work, we use the Keras library (22) within TensorFlow (23) and empirically optimize CNN parameters based on the training data (also see details in Discussion and Supplementary Table S2). Note that we followed the implementation and data preprocessing on the use of Convolution1D for text classification, available at https://github.com/keras-team/keras/blob/master/examples/imdb_cnn.py.
Method validity assessment
In the TPC task, we use precision, recall and the F-measure to calculate the performance score. Precision is the fraction of the number of relevant T2–T4 articles divided by the total number of predicted articles in this category. Recall is the fraction of the number of relevant T2–T4 articles divided by the number of actual T2–T4 articles in the gold-standard data set. The F-measure is the harmonic mean of recall and precision, which is calculated as follows: F-measure = 2 × [(recall × precision)/(recall + precision)].
We first perform cross-validation experiments on the training data set. The best-tuned model is then applied to the independent data in the test set.
Utility assessment
In addition to evaluating its validity, we also assess its utility in the real-world task of curating translational articles in PHGKB. In their routine workflow, human curators typically run eight PubMed queries (Supplementary Table S1) related to precision medicine twice a week and then manually examine all new search results before adding relevant ones to PHGKB. Moreover, they use PubMed's similar articles search to retrieve additional new candidate articles based upon a set of 283 translational research articles (https://media.nature.com/original/nature-assets/gim/journal/v19/n8/extref/gim2016210x1.doc) they previously identified (24). All new articles retrieved in both searches are then combined for a human review. Currently, the combined set of articles is sorted by their publication date during the human review. Alternatively, we propose to rank them by their likelihood to be translational articles, based on the predicted scores of CNN classifier. By doing so, we aim to establish a prioritized article review process to enable curators to focus on articles that are more likely to be relevant.
To test our hypothesis, we performed additional evaluation based on 2 weeks' worth of data in June 2017: from 6 June 2017 to 12 June 2017 (week 1), there are 1550 new articles collected in total and 62 are coded as T2–T4 articles by OPHG curators. Similarly, 1554 articles were retrieved between 22 June 2017 and 28 June 2017 (week 2) and 43 were coded as positive T2–T4 articles. Based on these data, we compute and compare the receiver operating characteristic (ROC) curve for two ranking strategies: sort by date versus by predicted relevance score of our classifier.
The 5-fold cross-validation results on training set
| Method | Precision | Recall | F1 |
|---|---|---|---|
| CNN | 0.7681 | 0.8785 | 0.8196 |
| SVM | 0.7688 | 0.7354 | 0.7517 |
Results
Method validity
As shown in Table 1, the CNN-based classifier achieves the best F-measure of 0.8196, with a higher recall than precision during the cross-validation experiments on the training set. Similarly, Table 2 shows the classification results on the independent test set. As can be seen, overall both classifiers yield slightly lower performances compared to the cross-validation results in Table 1. However, the CNN classifier still outperforms the SVM classifier consistently by a similar margin.
Method utility
As shown in Figure 2, the ROC curves indicate that our CNN classifier significantly outperforms the baseline method: it achieves an improvement of 29.6% and 28.6% for the 2 weeks, respectively. In addition, our analysis shows that on average in those 2 weeks, a curator only needs to review the top half of the papers when ranked by our CNN method in order to retrieve all relevant ones, suggesting an approximate saving of 50% in human curation time.
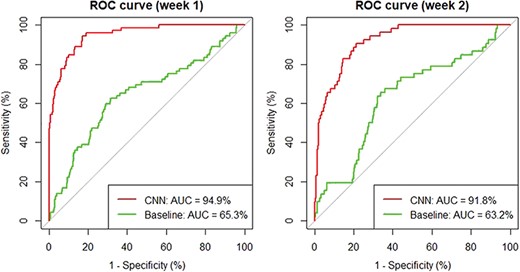
Comparing CNN with the baseline date-sort method using ROC curves.
Error analysis
In this work, we exploit machine learning to classify genomic research translational phase articles from the PHGKB and have achieved high classification accuracy overall. To better understand the computer predictions, we manually analyzed the classification errors of our CNN model on the test set, which includes 42 false positives (FPs) and 25 false negatives (FNs). The overall results are shown in Table 3. There are six FPs and one FN that were found to be incorrectly curated previously in the gold standard. There are also 12 borderline cases (7 in FPs and 5 in FNs) where the labels are somewhat ambiguous according to the curation criteria. Of the remaining 29 FPs, 5 articles are found to be case studies or meta-analysis of gene–disease associations (which are outside the scope) and 16 articles do not explicitly discuss human genetics or clinical utility. Of the 19 FNs, 2 articles do not have abstracts (i.e. only title information was used for the classification) and 10 articles describe pharmacogenomics, clinical screening or management (which are highly relevant to gene–disease associations).
The performance of the SVM and CNN models on the test set
| Method | Precision | Recall | F1 |
|---|---|---|---|
| CNN | 0.7614 | 0.8428 | 0.8000 |
| SVM | 0.7615 | 0.7232 | 0.7419 |
Statistics of FP and FN errors
| FPs | FNs | |
|---|---|---|
| Number of errors in the gold standard (mis-curated in the past) | 6 | 1 |
| Number of borderline articles (could be either T1 or T2 and above) | 7 | 5 |
| Number of mis-classified by our CNN method | 29 | 19 |
| Total | 42 | 25 |
Discussion
As mentioned earlier, for optimizing the CNN classifier performance, we empirically tested different parameters during the 5-fold cross-validation experiments on the training set. During the model training phase, its batch size controls the number of samples propagating through the CNN network. Hence, tuning the batch size affects the training loss curve and computation efficiency. Furthermore, the kernel size at convolutional layers defines the dimensions of feature maps, and this value affects how much the neighbor information can be processed. To obtain the best parameters of batch size and kernel size, we benchmarked with different combinations of the two parameters and subsequently analyzed the performance. We observed that the best value for the kernel size is 8 on our data. As for the batch size, we observed that the peak performance appears when it is between 50 and 80 (see Supplementary Table S2 also).
The classification task is quite challenging by nature, in that the number of translational articles is much smaller compared to basic research articles. To tackle the lack of positive training samples, interactive machine learning (25) might be worth exploring in the future. Since our CNN model uses word features to perform the classification task, it would be helpful to capture the important words involved in the machine decisions such that human experts can further analyze these word features and suggest additional improvements. A user-friendly visualization tool for neural networks can help in this regard. In the meantime, manual curators can also benefit from an explainable system (26) to feel more comfortable working with artificial intelligence (AI) algorithms.
Because the number of beyond bench-to-bedside phase publications is relatively small compared to initial discovery phase publications, it is difficult to further distinguish them into T2, T3 and T4 stages via machine learning, separately. This implies that a second level analysis with manual curation can differentiate the articles in T2, T3 or T4. Since there is a rapid increase in the number of publications within genomic research, the TPC task via machine learning would become complicated when an increasing proportion of genomic research publishes in later phases of translation.
In summary, our proposed approach using machine learning helps the horizon scanning to classify genomic translational research sufficiently, and a machine learning-based curation system is important to help curators successfully extract and address the later translational phases of genomic applications. We also expect that our approaches will decrease the published literature curation time to assist in the acceleration of genomic translational research. In our future work, we hope to clarify the influence of text features related to diseases and human genomics in the TPC task.
Funding
Intramural Research Programs of the National Institutes of Health, National Library of Medicine.
Conflict of interest. None declared.
References

{kind=link}
{kind=link}