





Abstract
Transcription factors (TFs) are DNA-binding proteins, which regulate many essential biological functions. In several cancer types, TF function is altered by various direct mechanisms, including gene amplification or deletion, point mutations, chromosomal translocations, expression alterations, as well as indirectly by non-coding DNA mutations influencing the binding of the TF. TFs are also actively involved in breast cancer (BC) initiation and progression. Herein, we have developed an open-access database, BC-TFdb (Breast Cancer Transcription Factors database), of curated, non-redundant TF involved in BC. The database provides BC driver TFs related information including genomic sequences, proteomic sequences, structural data, pathway information, mutations information, DNA binding residues, survival and therapeutic resources. The database will be a useful platform for researchers to obtain BC-related TF–specific information. High-quality datasets are downloadable for users to evaluate and develop computational methods for drug designing against BC.
Database URL: https://www.dqweilab-sjtu.com/index.php.
Introduction
Transcriptional regulation of genes is a vital process controlled by transcription factors (TFs) as key regulators to maintain cell homeostasis (1, 2). TFs regulate targeted gene expression by recognition and binding to specific DNA sequences known as transcription factor–binding sites (TFBSs) (3, 4). On the other hand, disruption of TF–target regulation leads to cellular damage and ultimately causes disease (4). Similarly, genetic alterations in cancer produce distinct tumor populations, which may remain benign or metastasize to distal sites (5, 6). For instance, heterogenic breast cancer (BC) tumor subtypes are underlined by a unique set of oncogenic alterations characterized in abnormal proliferation, invasion and metastatic potential (7). These genetic alterations are also co-related with drug resistance and relapse and offer attractive targets in therapeutic implications (8, 9).
TFs are also characterized in BC as tumor suppressors or oncogenes (10) and are key players in causing abnormal cellular growth (11). TFs also serve as important prognostic markers in BC. TF KLF4 (Kruppel-like factor 4) was reported as predictive pathological BC remission marker following neoadjuvant chemotherapy (12). Moreover, TFEts-1 expression was considered as an independent prognostic marker for recurrence-free survival (RFS) in BC (13). There is an increased interest in the identification of TFs as effective predictors for BC prognosis. Furthermore, it is also evident that tumor biomarker signature is crucial to explore more effective treatments for BC (8). Meanwhile, systematic studies to comprehensively visualize TF–target can be helpful to depict TF–target regulations. Integrated omics datasets for TFs (including TFBSs, target prediction, mRNA profiling and epigenetic status of chromatin) are considered as a useful resource for understanding TF–target regulations (14) and benefit researchers in transcriptional regulation studies (8).
In this study, we developed a database named BC-TFdb by utilizing a comprehensive approach to gather BC-related TF–target relationships. The resulting datasets in our study provide a comprehensive platform for studies related to TF–target regulation in BC. The BC-TFdb has integrated specific datasets with information from basic to advanced, reliable information to study TF–target regulations in BC. It will be helpful in providing online details to forecast potential co-association and co-regulation between TFs. In summary, BC-TFdb will serve as a useful resource for researchers to analyze TF regulation and gene expression in BC.
Methodology
Data collection and analysis
Bioinformatics tools have been widely utilized in large datasets to landscape genetic alterations in BC. Many groups identified these alterations through exome sequencing, copy number variation, mRNA and proteomic analyses of thousands of BC samples covering all major subtypes (9, 15, 16). Recently, whole-genome sequencing data of identified cancer-driving genetic alterations in 93 genes (17) and dataset of TFs relevant to RFS were also profiled (18). For database development, we collected human TFs associated with BC and involved in different BC pathways from different literatures (19, 20). Basic information regarding these TFs was also extracted from literature and used to divide into TF types. Genomic sequences (gene nucleotide sequences) and protein sequences of the collected set of TFs were retrieved from NCBI (National Center for Biotechnology Information) (https://www.ncbi.nlm.nih.gov/) (21) and UniProt platform (https://www.uniprot.org/) (22). Similarly, 3D structures for all data were downloaded from the PDB (Protein Data Bank) database (https://www.rcsb.org/) (23), and all the unavailable structures were submitted to Phyre2.0 for computational modeling (22, 24, 25). Information on the association of TFs in different BC-related pathways was obtained from KEGG (Kyoto Encyclopedia of Genes and Genomes) tool (26). In genetics, a missense mutation is a point mutation in which a single nucleotide change results in a codon that codes for a different amino acid. To obtain information on the missense mutations, the dbSNP (Single Nucleotide Polymorphism Database) database was accessed, and a list of mutations for each TF was retrieved. For screening these mutations, different tools such as SIFT (Scale-invariant feature transform) (27), PolyPhen (Polymorphism Phenotyping) (28) and CADD (Combined Annotation Dependent Depletion) (29) were utilized for prediction of deleterious nsSNPs (non-synonymous single nucleotide polymorphisms). SIFT tool (https://sift.bii.a-star.edu.sg/) determines the effect of amino acid substitution on protein’s function based on its physicochemical properties. SIFT scores nsSNPs as deleterious when the score is between 0 and 0.05, while nsSNPs with a score between 0.05 and 1 are declared as tolerated by SIFT (30). PolyPhen (http://genetics.bwh.harvard.edu/pph2/) is a web tool that forecasts the likely impact of an amino acid replacement on the structure and function of a human protein by combining physical and comparative parameters. Further, we also used CADD (https://cadd.gs.washington.edu/) to predict the most deleterious SNPs for each protein (TF) included. This framework integrates multiple annotations into one metric by contrasting variants that survived natural selection with simulated mutations. To understand the role of each TF in the initiation, progression and post-prognostic value survival analysis was performed using Kaplan–Meier plotter (31). Kaplan–Meier estimate is one of the best options to be used to measure the fraction of subjects living for a certain amount of time after treatment. Next, we also searched for the DNA-binding residues and designed a unique dataset included in our study design (32). For this purpose, we used DP-Bind (http://lcg.rit.albany.edu/dp-bind), which is a sequence-based approach for predicting the nucleic acid–binding residues in DNA-binding proteins (32). The server uses sequence evolutionary conservation information to predict the DNA-binding residues and their probability score. Finally, to address therapeutic implications, all the available drugs from PubChem (33) and DrugBank (34) were listed and included as a separate dataset for each TFs. Together these data will make a comprehensive platform for BC treatment options.
Development of database
Database development is an intricate task, as the designer has to accommodate the information that can be processed quickly, stored and responsive to the user needs easily. BC-TFdb offers large cloud-based online portals to ease data access and for in-depth analysis. BC-TFdb provides a friendly interface, allowing easy access to and efficient use of information. BC-TFdb search index was designed through various programming tools such as MySQL (Structured Query Language) (35), PHP (Hypertext Preprocessor) (36), AJAX (Asynchronous JavaScript And XML) (37), HTML (Hypertext Markup Language) (38), jQuery (39) and Bootstrap.
Back end preparation
Database designing was done using WAMP server. Open access was ensured by scripting in environments like HTML and PHP. Data storage, manipulation and retrieving from the databases were managed through MySQL. WAMP server provides an environment to create a web application with PHP, Apache and MySQL database and is equipped with PHP MyAdmin to confer full control over the web contents.
Front-end preparation
The database interface needs intelligence as the users interact directly with it and increase its overall acceptance. Client-side or front-end design aims to deliver an attractive interface to accomplish tasks efficiently and easily without having much of bioinformatics knowledge of command-line interface. Popular tools such as Bootstrap, CSS (Cascading Style Sheets) and HTML were used to tailor BC-TFdb. CSS is a high-level programming language commonly used to customize and style PHP and HTML script. The database provides a user-friendly platform for easy access and operation of TFs related data. The overall workflow of the work, including the data source and other information, is given in Figure 1.
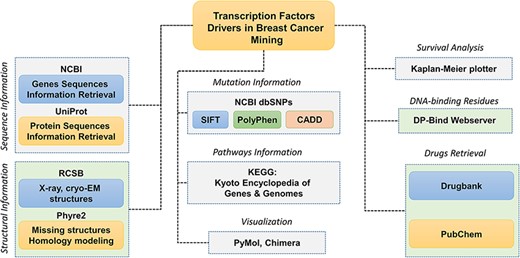
Schematic representation of the database components and data analysis modules used to obtain the specific results. Each tab is tagged, and their respective analyses of webservers or software are also given.
Results and discussion
BC-TFdb primarily focuses on the multiple features regarding the TFs in BC. This database is a collective platform for a total of 161 TFs involved in BC progression. The database includes multiple tabs including basic information, sequence and structural information, pathway information, survival information, DNA-binding residues, missense mutations and their impact, and therapeutics for the representation of a specific type of data. The overall workflow of the strategy used in the design of this database has been given in Figure 1.
Structure and implementation of the database modules
Different modules have been used for designing the BC-TF-specific database and are discussed as follows.
Basic information tab
The basic information page represents a sheet that contains details about the genes and proteins, UniProt entry ID, organism type, length, KEGG accession IDs and string PPI (protein-protein interactions) accession number. This tab is also equipped with the search module for accessing any specific information about the TF of interest. This information will help the user to access specific information about each TF. The representative image of this tab is presented in Figure 2.
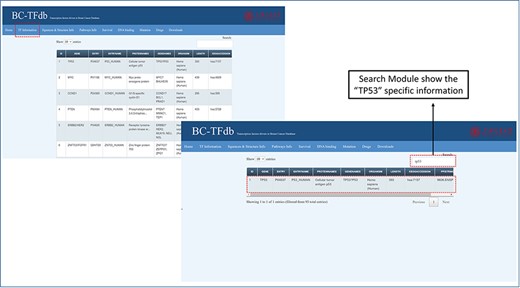
Representative image of the basic information tab showing details about TFs from the dataset of the cohort study. The bottom panel shows the search module implemented in the database to obtain specific information.
Sequence and structural information tab
Genomics, proteomics and structural information retrieved from UniProt, NCBI, RCSB (Research Collaboratory for Structural Bioinformatics), Ensembl and gnomAD are all available in this tab. This tab is further divided into three sub-modules, which represent gene sequences, protein sequences and structural information separately. The first sub-tab represents the gene sequences along with the gene names and nucleotide lengths. Similarly, the proteomics tab represents the protein sequences with the amino acid length and protein name. Users can access specific information on both genomics and proteomics by searching using the sequence features or gene name in the search engine. Additionally, the structural information tab provides data on these TFs. This sheet provides information about the gene name, PDB entry ID (RCSB), structural determination method (X-ray, NMR (Nuclear magnetic resonance), cryo-EM (Electron Microscopy) or homology model), resolution of the structure and chain information. This sheet also owns information about the AA position of the specific structure. Moreover, the download module allows users to easily download the specific structure. Furthermore, 14 unavailable structures were modeled using Phyre2 homology modeling server and included as downloadable information deposited in the online database. The representative image tutorial of this tab is presented in Figure 3.
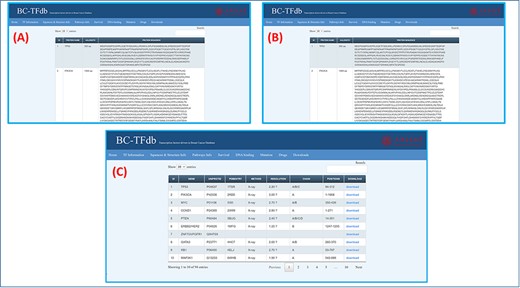
Representation of the Sequence and structural information tab. (A) shows the tutorial image for accessing the gene sequence information of TFs. (B) shows the tutorial image of accessing the protein sequence information of TFs. (C) shows the tutorial image of accessing the protein structural information of TFs.
Pathway information tab
For pathway information about specific TFs, KEGG was accessed to retrieve listed information about the involvement of the TFs in BC pathways. This information was deposited on the pathway information tab for easy access to users. Specific pathway information can be viewed and downloaded as an image file. Further, the search module can also be utilized to retrieve the pathway-specific information based on the gene name. The image tutorial of the pathway information tab is represented in Figure 4.
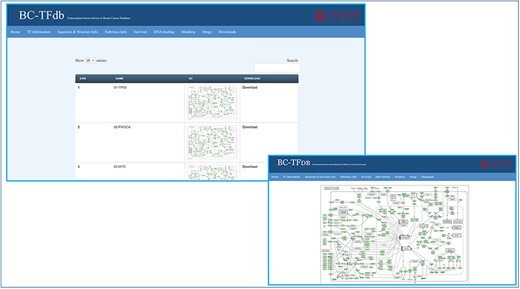
Representative tutorial image of the Pathway information tab upon online access. The bottom panel views a sample pathway of TP53.
Survival information tab
The database also includes survival information about each TF included in the study related to BC. Users can easily retrieve the TF-specific survival information such as P value and hazard ratio. Moreover, the sheet also has the gene name and Affymetrix ID of each TF. The specific information can be searched in the search module either using the gene name or Affymetrix ID. Furthermore, with the downloads module, user can also download the survival plot of each TF available in this tab. KM plotter was utilized for the survival analysis of BC-specific TFs, and survival plots for each of the TF were searched, retrieved and deposited here. The image tutorial of the Survival information tab is represented in Figure 5.
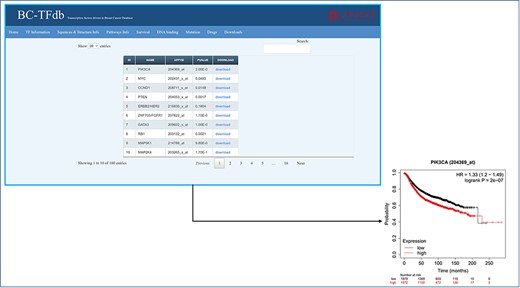
Representative image of the Survival information tab, including a list of all TFs and their downloadable KM (kaplan-meier plot) plots. The bottom panel shows the sample survival KM plot for PIK3CA.
DNA binding tab
Information about the DNA-binding residues is crucial to understand the role of specific residues in the recognition of DNA or RNA by the TFs, which may help in the BC diagnosis and therapeutic development. These hotspot residues may also serve as drug hotspots for binding interactions and novel drug development. On the other hand, lack of such information hinders the exploration of the molecular mechanism of DNA or RNA recognition. It, thus, limits the understanding of cancer prognosis and development at a molecular level. No single platform is available to provide information about the role of specific residue in the DNA or RNA recognition. Thus, we used DP-Bind to predict the DNA-binding residues of each TF. The server predicts DNA-binding residue represented with 1 for binding and 0 for nonbinding residues. Herein, information about the DNA-binding residues of each TF is given. Only residues that were predicted to be DNA binding are given in the sheet. A total of 6180 residues for all the TFs are predicted to be DNA binding, while the rest were discarded as nonbinding residues. The DNA binding tab provides information about the protein name, residue position, residue name, SVM (support vector machine) binding score and SVM probability. The DNA-binding residues of each TF are arranged according to the highest SVM probability values. The search module using the gene name displays gene-specific DNA-binding residue information. Further, these analyses would also help the experimental researchers to validate the binding probability of each residue for a specific TF. The image tutorial of the DNA binding tab is represented in Figure 6.
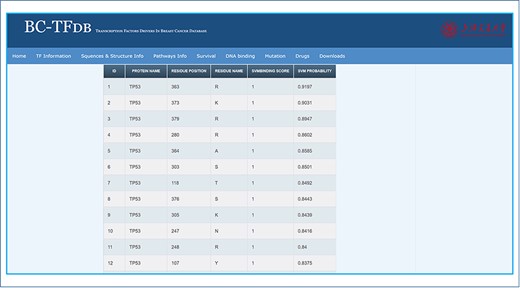
Representative image of the DNA binding tab, including a list of all TFs and listed SVM scores.
Mutations tab
Mutations are highly correlated with the complex disease phenotype and, thus, of great concern in cancer research. Any substitution in the protein-coding region may alter the protein structure and function. Therefore, depicting the impact of each mutation may help to understand the disease initiation and progression. Similarly, TF-specific mutations contribute as the driving force in BC progression. Moreover, the identification and characterization of these mutations are crucial to better understand the underlying mechanisms in BC development. This also offers baseline data to design improved therapeutic strategies in BC. Herein, we listed all the possible SNPs in a single tab with a large dataset, including each of the TF. The non-synonymous mutations of each TF were retrieved from dbSNP database and processed using SIFT, PolyPhen and CADD servers. These servers help to predict and classify the mutation class, i.e. deleterious, tolerated, benign, possibly damaged or likely benign based on the specific scores. A total of 163 datasets were processed, which contain 142 435 mutations in total and grouped based on the specific output. This mutation-specific information about each TF is provided to users for retrieval. The mutations tab is featured with different kinds of information including gene name, variant ID, mutation class, consequence type, AA name, AA co-ordinates, SIFT sort, SIFT class, SIFT score, PolyPhen sort, PolyPhen class, PolyPhen score, CADD sort, CADD class and CADD score. The specific mutation tagged with specific variant ID is classified as deleterious or tolerated by the SIFT server based on the SIFT score, benign or probably damaging based on the PolyPhen score by the PolyPhen server and likely benign or likely deleterious by the CADD server based on the CADD score. The implemented search module retrieves mutation information based on gene name or variant ID. This module may help the researchers to directly access the impact of a specific mutation in these TFs. The image tutorial of the Mutations tab is represented in Figure 7.
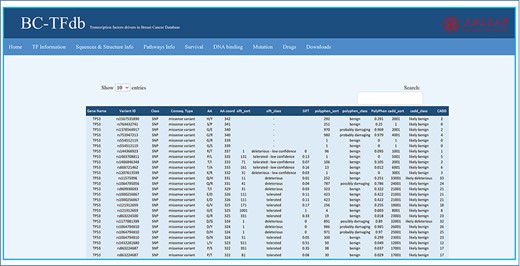
Representative image of the Mutations tab with various headers representing each feature.
Drugs tab
Therapeutic development of potential drugs or vaccine candidates is important for disease curtailment. The development of drugs against cancer disease is hot research and needs further investigation. Drug repositioning may be of great interest in research to discover novel drug–target interactions. For this purpose, knowledge about previous drugs and their mode of action is important. Hence, to provide information about the available drugs for these TFs’ drug target, the drugs tab was integrated as a part of this database. In this tab, information regarding the target name, DrugBank ID, drug group, pharmacological action and molecular action are provided. The sheet contained information about the DrugBank accession ID of each drug, the drug name and their respective targets. Further, these drugs are divided as either experimental, investigational or approved. Furthermore, information about the pharmacological action, whether the mechanism is known or unknown, are also provided. The classification of each drug as an inhibitor, downregulator, antagonist, agonist, chaperon, binder, inducer, substrate or antibody is also provided in the Actions heading. This information about the drugs of each TF may help the user to retrieve it easily for therapeutic repurposing. The image tutorial of the Drugs tab is represented in Figure 8.
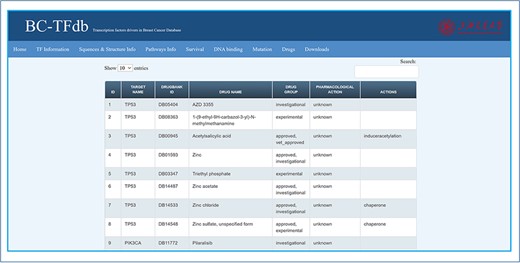
Representative image of the Drugs tab with various headers representing each feature.
Downloads tab
The Downloads tab enlists all the data as downloadable features. All the data included in the database could be downloaded as a separate file. The image tutorial of the Downloads tab is represented in Figure 9.
Representative image of the Downloads tab that contains all the files for downloads.
Conclusion
In conclusion, the BC-TFdb provides a comprehensive platform for research, to easily access and retrieve information related to TF drivers in BC. The platform provides useful information including gene and protein sequences, protein 3D structures, BC pathway information, survival information, DNA-binding residues, missense mutations and their impact and therapeutics such as the available drugs used against the specific TF targets. This platform may help the researcher to access BC-specific information for computation and experimental analysis. This platform would help to better understand the role of each TF in pathogenesis and would map a better strategy for the management and therapeutic feasibility of BC.
Acknowledgements
The computations were partially performed at the PengCheng Laboratory and the Center for High-Performance Computing, Shanghai Jiao Tong University.
Funding
National Science Foundation of China (32070662, 61832019, 32030063), Key Research Area Grant 2016YFA0501703 of the Ministry of Science and Technology of China, the Science and Technology Commission of Shanghai Municipality (19430750600), as well as SJTU (Shanghai Jiao Tong University) JiRLMDS Joint Research Fund and Joint Research Funds for Medical and Engineering and Scientific Research at Shanghai Jiao Tong University (YG2021ZD02) to D.Q.W. National Natural Science Foundation of China (31801037) and Science Foundation of Army Medical University (2017XQN01, 410310543403, 2019JCZX05) to X.Z.
Conflict of interest.
None declared.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}