





Abstract
The power of next-generation sequencing has resulted in an explosive growth in the number of projects aiming to understand the metagenomic diversity of complex microbial environments. The interdisciplinary nature of this microbiome research community, along with the absence of reporting standards for microbiome data and samples, poses a significant challenge for follow-up studies. Commonly used names of metagenomes and metatranscriptomes in public databases currently lack the essential information necessary to accurately describe and classify the underlying samples, which makes a comparative analysis difficult to conduct and often results in misclassified sequences in data repositories. The Genomes OnLine Database (GOLD) (https:// gold.jgi.doe.gov/) at the Department of Energy Joint Genome Institute has been at the forefront of addressing this challenge by developing a standardized nomenclature system for naming microbiome samples. GOLD, currently in its twenty-fifth anniversary, continues to enrich the research community with hundreds of thousands of metagenomes and metatranscriptomes with well-curated and easy-to-understand names. Through this manuscript, we describe the overall naming process that can be easily adopted by researchers worldwide. Additionally, we propose the use of this naming system as a best practice for the scientific community to facilitate better interoperability and reusability of microbiome data.
Introduction
We live in a microbial world, surrounded by trillions of bacteria, viruses and fungi. The human body is estimated to contain more microbes than the actual number of human cells (1). This diverse collection of microbes that naturally live in any habitat including our body and its surrounding environment is commonly referred to as the microbiome. Recent advances in high-throughput sequencing methodologies have rapidly expanded our ability to explore the microbiome and resulted in the exponential growth of a number of metagenomic and metatranscriptomic studies. The characterization of microbial communities with a goal to understand how they interact with each other in their natural ecosystems and own environments is a central task for such studies. As a result, we have significantly progressed in surveying various biomes, being able to dissect microbial communities to strain-level resolution without the need to culture individual organisms (2). Analyses of metagenomes have led to a comprehensive understanding of complex microbial communities from a variety of environments (3–5). The emerging metagenomic next-generation sequencing has allowed researchers to provide a comprehensive clinical diagnosis to identify pathogenic organisms in infectious diseases (6).
To better understand the complexity and composition of environmental microbiomes and to compare with related environments, it is extremely important to capture the full contextual information of the sample being sequenced. There have been global efforts such as the implementation of ‘environmental packages’ established by the Genomic Standards Consortium to capture metadata in a standardized way (7). Despite such initiatives, the number of metadata associated with a particular sample or organism can vary widely. This lack of metadata makes analysis tasks difficult because researchers cannot accurately extract and interpret all the necessary information from the samples they analyze. While isolate genomes have established principles of phylogeny that help with comparative analysis when additional metadata is missing, metagenomes lack a systematic classification, and there are no specific guidelines to name them even when they are deposited in public databases like National Center for Biotechnology Information (NCBI). Because of this, samples coming from a similar environment may have very different names. Researchers often decide to name their samples using cryptic, esoteric names with labels or identifiers from their laboratory notebooks. This makes it arduous and often impossible for others to look at the name of the sample and get a sense of where it is coming from and how it is related to other samples. For example, if one is analyzing freshwater lake samples from around the country and names their metagenomes ‘aquatic’, it will be impossible to identify the source of the sample because it could come from an ocean, a river or drinking water. Therefore, there is a pressing need for a standardized naming system for microbiome samples.
Genomes OnLine Database (GOLD) is a manually curated microbiome metadata management system at the Department of Energy Joint Genome Institute (JGI) (8). GOLD hosts hundreds of thousands of projects and associated metadata from around the world, including >180 000 public metagenomes and metatranscriptomes. All of these projects are manually curated by GOLD staff using a canonical naming system (9) that has been standardized and practiced for >12 years. Sequence data linked to the GOLD curated projects get annotated and are made available through the Integrated Microbial Genomes and Microbiomes portal (10) for comparative analyses. GOLD projects and metadata are used in shareable, reproducible workflows called Narratives within the KBase data science platform (11) and are also used by MGnify (12) curators to add supplementary metadata to projects imported from the European Nucleotide Archive (13). In this paper, we describe GOLD’s naming process in detail with specific examples of samples from various environments. Additionally, through this manuscript, we encourage the broader microbiome research community to adopt this naming system and use it in publications and for depositing data to public repositories.
GOLD’s organization and project sources
GOLD is organized into a four-level classification system consisting of studies at the top, and under them, there are organisms or biosamples. An organism represents a cultured or uncultured taxonomic entity, whereas a biosample represents a microbiome sample collected from a given environment. At the third level, we have sequencing projects; they are either isolate genome projects connected to individual organisms or metagenome or metatranscriptome projects connected to biosamples. Analysis projects at the fourth level represent the set of analyses done on the sequence data generated on the projects. Thus, one can visualize the GOLD entities organized hierarchically into study → organism/biosample → sequencing project → analysis project.
There are three main sources from where projects are added to GOLD: (i) projects from samples sequenced at JGI as part of its several user programs, (ii) those imported from public repositories such as NCBI and (iii) projects submitted by external users. When metagenomes are sequenced at JGI or imported from NCBI, their corresponding biosamples’ names are curated by GOLD curators according to the provided metadata. For NCBI metagenomes, when sufficient metadata is not readily available to construct the name, curators may have to refer to the available publications associated with the NCBI BioProjects. Table 1 lists a few examples to demonstrate how originally submitted cryptic sample names were curated in GOLD.
Examples of microbiome samples from JGI and NCBI with names before curation and after applying GOLD’s standardized naming conventions
| Submitted name | GOLD’s curated name |
|---|---|
| JGI projects | |
| 003-ER18-SC-SDNA | Soil microbial communities from watershed of Upper East River, CO, USA - 003-ER18-SC-SDNA |
| L5_T2_FL_03 metagenome | Sugarcane leaf microbial communities from experimental field in the University of Florida, Reddick, FL, USA - L5_T2_FL_03 |
| UBC_AA-12-1 | Anaerobic digester fluid microbial communities from the University of British Columbia, Vancouver, Canada - UBC_AA-12-1 |
| WIN1-9-19-19 metagenome | Sediment microbial communities from irrigation canal in Merced County, CA, USA - WIN1-9-19-19 |
| CRU5 metagenome | Spruce roots microbial communities from Bohemian Forest, Czech Republic - CRU5 |
| Y16_303_L2_tf | Freshwater microbial communities from Lukens Lake, Yosemite National Park, CA, USA - Y16_303_L2_tf |
| NCBI projects | |
| qiita_sid_1711:1711.KAJ1.1 | Agricultural soil microbial communities from a farm in Kakamega, Kenya - 1711.KAJ1.1 |
| 1521.EB064.s.6.1.sequences | Sediment microbial communities from Toolik Lake, AK, USA - 1883.2008.276 |
| 11 116.L01A078.1194251 | Maize rhizosphere microbial communities from Lansing, MI, USA - 11 116.L01A078.1194251 |
| EMOSE_N010000374 | Seawater microbial communities from epipelagic zone of the Mediterranean Sea - EMOSE_N010000374 |
| pGvST_028_VLP | Human feces viral communities from FMT recipient with GvHD at the Prince of Wales Hospital, Shatin, Hong Kong - pGvST_028_VLP |
| TBL78 | Leaf surface microbial communities from Ti plant at Nanyang Technology University campus, Singapore - TBL78 |
GOLD’s canonical naming process
The naming of a metagenome or a metatranscriptome project begins with naming a biosample associated with it. However, before describing GOLD’s standardized naming system, we would like to emphasize that the concept of a biosample in GOLD is slightly different from the concept being used for NCBI’s BioSamples (14). The GOLD biosample concept is exclusively applied to describe the environment from where a metagenomic or metatranscriptomic sample was collected. Thus, the GOLD biosample represents only the metadata that is associated with a particular microbiome sample. This is in contrast to an NCBI BioSample, which is applicable to both environmental/microbiome samples and organisms.
To create a canonical name for a biosample, GOLD uses four distinct types of metadata to systematically construct a standardized name (Figure 1). They are habitat, type of communities, detailed location and an identifier. (i) The habitat indicates a specific environment from which the sample was collected. (ii) The type of communities indicates the targeted group of organisms in the physical specimen; such group can be broad: microbial or eukaryotic, or more specific: bacterial, archaeal, viral or fungal. (iii) The location contains (but not limited to) the information about a specific geographic location, including a country or an ocean, of the sampling site. (iv) The identifier, which is unique to each biosample, is usually a combination of letters, numbers and/or special characters to distinguish a biosample from the closely related ones.

A schematic representation of a standardized biosample name with its constituent components of habitat, community, location and identifier.
The very first metadata type, habitat, is ecological metadata. By definition, a habitat is a specific environment, a combination of biotic and abiotic factors, where targeted organisms live. That is why the canonical naming is closely related to GOLD’s five-tiered ecosystem classification, which consists of controlled vocabulary terms divided among ecosystem, ecosystem category, ecosystem type, ecosystem subtype and specific ecosystem categories (15). The first tier of the GOLD ecosystem classification is divided into environmental, host-associated and engineered and aims to capture the broader environment from which the sample originated from. Each of these three divisions has further broken down categorically all the way to the fifth level or specific ecosystem. Figure 2 gives an overview of the ecosystem distribution of >180 000 public biosamples in GOLD until the third tier or ecosystem type. For example, ∼80 000 biosamples in GOLD are categorized as environmental, out of which 60% are aquatic, 38% are terrestrial and 2% fall under air ecosystem category. The aquatic samples are further classified into multiple GOLD ecosystem types: marine, freshwater, thermal springs, etc.
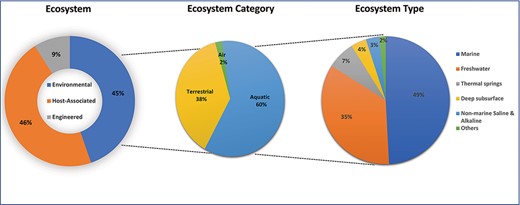
The distribution of 180 677 public biosamples among the top three GOLD ecosystem classification levels.
Based on the metadata availability, individual users have a lot of flexibility when it comes to deciding on the habitat (beginning of their biosample name) of their sample. A habitat can include very specific terms from GOLD’s specific ecosystem (fifth tier), e.g. ‘sediment’, ‘microbial mats’, ‘feces’, ‘leaf surface’, etc. Or, in some cases, they can be a little more generic, similar to GOLD’s ecosystem subtype (fourth tier) terms, such as ‘rhizosphere’, ‘gills’, ‘soil crust’, ‘microbialites’ or even similar to GOLD’s ecosystem type (third tier) with terms like ‘roots’, ‘freshwater’, ‘marine’ or ‘soil’. This can happen when there is not enough metadata for a particular sample. For example, if a user is unsure about the source of the soil sample (forest, agricultural, desert, etc.), they can name a biosample as: ‘Soil microbial communities from Concord, CA, USA - S1.’ Alternately, if the soil sample is from the forest soil, specifically from The Giant Forest at Sequoia National Park, then a biosample name such as ‘Soil microbial communities from Giant Forest, Sequoia National Park, CA, USA - S1’ becomes a lot more informative. In some cases, a habitat can even include a combination of two separate ecosystem tiers to make it more specific: ‘freshwater sediment’, ‘root nodules’ or ‘cave wall biofilm’ to name a few. The biosample’s name structure may slightly vary from one ecosystem to another. In the following section, we provide examples of canonical naming applied to samples from all three types of ecosystems.
GOLD’s canonical naming of biosamples from various ecosystems
Environmental biosamples
Environmental samples are the ones that were collected from environmental ecosystems: aquatic, terrestrial or air. Currently, there are >91 500 environmental biosamples in GOLD. Among them, there are ≥55 000 aquatic biosamples, ≥34 700 terrestrial biosamples and >1650 air biosamples. Each environmental biosample has a habitat associated with a specific environmental ecosystem category and ecosystem type: soil, freshwater, seawater, marine sediment, indoor air, etc. So, how does the naming process for these biosamples work in practice?
Let’s say, someone wants to examine fungal communities of a physical sample collected from forest soil in the Sierra National Forest in California and marked as ABC_1 in their laboratory journal. In this case, the canonical name of the biosample in GOLD can be ‘Soil fungal communities from the Sierra National Forest, CA, USA - ABC_1’, where ‘soil’ is a habitat, ‘fungal communities’ are targeted organisms, ‘the Sierra National Forest, CA, USA’ is the location and ‘ABC_1’ is the identifier.
Here are examples of the GOLD canonical biosamples’ names for various environmental samples: ‘Soil microbial communities from paddy field in Yamagata Integrated Agricultural Research Center, Japan - YGP1’; ‘Seawater microbial communities from Station 08, Station ALOHA, North Pacific Gyre, Pacific Ocean - CSHLIID20-03a-S08C001-0015’; ‘Sediment microbial communities from a vernal pool in Lake County, CA, USA - SR-VP_26_10_2019_B_35cm’; and ‘Freshwater microbial communities from Blue River watershed, KS, USA - 0WARd229’.
Host-associated biosamples
Another large group of biosamples in GOLD (∼88 500) fall under the host-associated category. The biosamples from this group are associated with various host organisms, such as animals, plants, fungi, protists or prokaryotes. Additionally, they come from various biological tissues or body products of their respective hosts, like feces, blood, roots, leaves, mycelium, etc. All of this information is reflected in the biosample name. Since these samples come from specific host organisms, the biosample name should incorporate the name of the host. In GOLD, a common organism name is generally used in a biosample name in order to facilitate the easier searching of projects and samples from similar host organisms. Let’s say someone decided to examine dog’s saliva viruses from a shelter in Walnut Creek, California, and collected physical samples from three dogs, two of which were sick and one was healthy. The researcher may have named the samples in their laboratory notebook as ‘BGS’ (sick dog 1), ‘BGH’ (healthy dog) and ‘GSS’ (sick dog 2). When the names of the respective biosamples are curated in GOLD, the final result will be as follows:
‘Dog saliva viral communities from a sick animal in ARF shelter, Walnut Creek, CA, USA - BGS’; ‘Dog saliva viral communities from a healthy animal in ARF shelter, Walnut Creek, CA, USA - BGH’; and ‘Dog saliva viral communities from a sick animal in ARF shelter, Walnut Creek, CA, USA - GSS’.
The following are a few more examples of curated names for host-associated biosamples in GOLD:
‘Human feces microbial communities from FMT recipient with GvHD at the Prince of Wales Hospital, Shatin, Hong Kong - pGvST_011’; ‘Blood plasma viral communities from a lung transplant patient, the Medical University of Vienna, Austria - S133_2’; ‘Komodo dragon skin microbial communities from Fort Worth, TX, USA - 207507.10512.skin’; ‘Bee gut microbial communities from Hubei, Yichang, China - bee10.3’; ‘Rice rhizosphere microbial communities from paddy field in Mishima, Shizuoka, Japan - 1642.MS00512’; and ‘Chive leaf microbial communities from plant growth chamber in Institute of Urban Environment, CAS, Xiamen, China - PCM3’.
Engineered biosamples
In GOLD, an engineered ecosystem is any man-made/artificial environment that includes but is not limited to bioreactors, wastewater treatment plants (WWTPs), mesocosms, built environments, industrial products, etc. There are >18 000 such biosamples in GOLD.
Let’s say someone wants to examine microbial communities in wastewater effluent collected as three biological replicates from the Columbia Boulevard WWTP in Portland, Oregon, that are labeled as WE_1, WE_2 and WE_3 in their laboratory notebook. In this case, the submitter needs to create three distinct biosamples in GOLD and name them as ‘Wastewater effluent microbial communities from Columbia Boulevard WWTP in Portland, OR, USA - WE_1’; ‘Wastewater effluent microbial communities from Columbia Boulevard WWTP in Portland, OR, USA - WE_2’; and ‘Wastewater effluent microbial communities from Columbia Boulevard WWTP in Portland, OR, USA - WE_3’.
The following are a few more examples of curated names for engineered biosamples in GOLD:
‘Swine feedlot wastewater viral communities from WWTP in Huizhou, China - Aerobic_3’;
‘Biofilm microbial communities from reverse osmosis membrane in Water Desalination and Reuse Center, KAUST, Thuwal, Saudi Arabia - U1.1’; ‘Sauerkraut microbial communities from San Diego, CA, USA - food.sourkraut.2.1.2.H’; and ‘Mesophilic digester sludge microbial communities from biogas plant in Geslau, Bavaria, Germany - PB25_MT_150518’.
Special cases
As evident from the examples given earlier, the canonical naming of the biosamples is a relatively straightforward process. However, there are some samples that need special attention.
Enrichments
Sometimes, after collecting a sample, a researcher enriches it to propagate a specific group of organisms, e.g. methane-oxidizing bacteria or carbohydrate-degrading communities. Often, during the enrichment process, they collect several samples at specific timepoints. In such cases, it is recommended that the words ‘lab enriched’ is added at the beginning of the name, while the details about the enrichment process and timepoints can be added to the biosample description. The following are a couple of examples of the names of such biosamples: ‘Lab-enriched seawater microbial communities from Canoe Beach, Nahant, MA, USA - OXC2017-Chitosan204’ and its description: ‘Enriched seawater microbial communities from Canoe Beach, Nahant, MA, USA; seawater bacteria enriched on chitosan beads sampled in 204 hours of enrichment’. Biosample name: ‘Lab-enriched soil microbial communities from grassland in Hebei, Zhangjiakou, China - CK60.2’ and description: ‘Lab enriched soil microbial communities from grassland in Hebei, Zhangjiakou, China; un-amended soil spiked with tetracycline, chlortetracycline and oxytetracycline, each at 150 mg/kg dry soil and incubated at 25°C in the dark for 60 days’.
Cocultures and contaminated cultures
Cocultures and contaminated cultures are treated as metagenomic/microbiome samples in GOLD. In the case of cocultures, all one needs to do is to add the word ‘coculture’ either in front or after the name(s) of the major organism(s) in the culture. ‘Coculture of unialgal cyanobacterium with coexisting heterotrophic bacterium from Dresden University of Technology, Germany - Calothrix_61.4/Streptobacillus’; ‘Roseovarius nubinhibens ISM and Alexandrium tamarense coculture from University of Georgia, USA - T8_LowN_B’. The name of a biosample from a contaminated culture can be constructed the similar way: ‘Contaminated culture of Pseudomonas putida S.12 from the Institute of Biology Leiden, Netherlands - S12Pp’; ‘Contaminated culture microbial communities from University of Georgia, Athens, GA, USA - Monilinema alkalinum CCIBt3284’; and ‘Contaminated culture fungal communities from Oregon State University, United States - Rhizopus stolonifer NRRL 66455’.
Conclusion and future plans
In this article, we have demonstrated (i) the need as well as the importance of a standardized microbiome sample naming that is self-explanatory without digging deep into buried structured metadata and (ii) the ease of the uniformly applying naming convention to samples coming from different streams. To date, we have curated ∼180 000 microbiome samples that have come from various streams, including public repositories like NCBI. We have applied standardized naming conventions to samples submitted to JGI for sequencing, and we have deposited these standardized names to the NCBI BioSample database (14). Through help documents and individualized user support, GOLD ensures that all externally submitted microbiome samples adhere to the standardized naming conventions. Thus, we have practiced this naming convention and demonstrated that it can be applied to data coming from different streams, i.e. from public repositories, direct submissions by users and the projects carried out in-house at JGI. Now, we aim to promote and extend this microbiome naming convention to a wider community by undertaking the following steps:
(i) Reach out to the microbiome research community through this publication to adopt this naming approach. We aim to provide help documents and video training for users to understand and adopt standardized naming conventions.
(ii) Conduct one or more workshops on canonical naming approaches, so that researchers can familiarize, adopt and use this naming convention. Through these workshops and community outreach programs, we aim to seek input and improve the usability of our naming conventions.
(iii) Communicate with microbiome data repositories like NCBI, National Microbiome Data Collaborative and MGnify (12, 16, 17) and coordinate to help their users in adopting the canonical naming standards.
Funding
The work conducted by the U.S. Department of Energy Joint Genome Institute (https://ror.org/04xm1d337), a DOE Office of Science User Facility, is supported by the Office of Science of the U.S. Department of Energy operated under Contract No. DE-AC02-05CH11231. This work used resources of the National Energy Research Scientific Computing Center, which is supported by the Office of Science of the U.S. Department of Energy.
Conflict of interest
None declared.
Acknowledgements
We thank the microbiome user community who has been submitting samples to GOLD and worked with us in adopting the naming convention and providing feedback to improve it over a period. We also would like to thank Jagadish C. Sundaramurthi for the initial discussions about this manuscript.

{kind=link}
{kind=link}