





Abstract
The big-data analysis of complex data associated with maize genomes accelerates genetic research and improves agronomic traits. As a result, efforts have increased to integrate diverse datasets and extract meaning from these measurements. Machine learning models are a powerful tool for gaining knowledge from large and complex datasets. However, these models must be trained on high-quality features to succeed. Currently, there are no solutions to host maize multi-omics datasets with end-to-end solutions for evaluating and linking features to target gene annotations. Our work presents the Maize Feature Store (MFS), a versatile application that combines features built on complex data to facilitate exploration, modeling and analysis. Feature stores allow researchers to rapidly deploy machine learning applications by managing and providing access to frequently used features. We populated the MFS for the maize reference genome with over 14 000 gene-based features based on published genomic, transcriptomic, epigenomic, variomic and proteomics datasets. Using the MFS, we created an accurate pan-genome classification model with an AUC-ROC score of 0.87. The MFS is publicly available through the maize genetics and genomics database.
Database URL https://mfs.maizegdb.org/
Introduction
The study of cellular, molecular and genetic interactions in maize generates huge amounts of data. Due to the high dimensionality and heterogeneity of multi-omics data, integrating and analyzing these datasets has proven to be extremely difficult. Recently there has been an increased interest in analyzing large-scale omics data, particularly for predicting genotype-phenotype relationships. Over the last decade, machine learning has found numerous applications in plants, resulting in a slew of papers and reviews (1–3). There has been particular interest in maize, making it the most studied crop using machine learning (4). This interest can be attributed to the fact that it is grown in many parts of the world and has a variety of uses, including direct human consumption, animal feed, the production of ethanol and other biofuels.
To further advance and facilitate the application of machine learning in crop and plant research, robust analytical methods and tools are required to manage multi-omics data through efficient data management, linkage and integration strategies. This need is particularly strong for maize research, where a vast amount of data exists. Numerous storage methods have been developed to manage and analyze multi-omics data (5), including the Maize Genetics and Genomics Database (MaizeGDB) (https://www.maizegdb.org/), which comprises maize reference sequences, diversity data, expression data, phenotypic data, epigenetic and regulatory data, as well as metabolic pathway data along with multiple tools for genome-wide maize data exploration (6); Panzea (https://www.panzea.org/), comprising genotypic and phenotypic data from several maize lines (7); and Phytozome (https://phytozome-next.jgi.doe.gov/) a centralized hub of annotated plant gene families, evolutionary data and functional data (8). Other comprehensive databases and data repositories such as GenBank (https://www.ncbi.nlm.nih.gov/genbank/) (9), Gramene (http://www.gramene.org/) (10), ePlant (http://bar.utoronto.ca/eplant_maize/) (11), MODEM (http://modem.hzau.edu.cn/) (12) and a more recent maize multi-omics database ZEAMAP (http://www.zeamap.com/) (5) also collect maize omics data. While these databases are quite useful, they store data in a structured manner using relational databases and require advanced multi-layer data structures to optimize data management and analysis. Additionally, they frequently lack interactive multivariate methods for exploring and integrating datasets. These databases enable users to access data in various file formats, including annotation data in GFF format and SNP datasets in VCF format. Although these datasets are easily accessible via these repositories, they do not come in a format suitable for performing diverse multivariate analyses, particularly at the gene level. Users who wish to apply modeling to these multi-omics datasets must spend considerable time collecting, cleaning and aggregating before using them for model training.
Regardless of the challenges, omics integration studies have pervaded literature in recent years (13–15). As a result, the growing collection of omics data in maize is gaining attention among researchers to carry out systematic integrative analysis and storage of the heterogeneous data (16). In response to the challenges of handling heterogeneous data, non-traditional databases (NoSQL) emerged as an alternative, more flexible and more scalable data store (17, 18). Therefore, this paper presents the Maize Feature Store (MFS), a NoSQL-based interactive, modular and dynamic user interface for systematically integrating and analyzing over 14,407 gene-based features based on the most recent maize multi-omics dataset (version 5 of the B73 reference genome, or B73v5). Feature stores are becoming a powerful resource for data scientists to have readily available access to high-quality features for rapid deployment of machine learning applications, but feature stores are not available for most model organism databases. We aim to demonstrate how MFS provides a suite of methods and modeling modules, enabling users to find meaningful patterns from the maize omics data.
To demonstrate the utility of the MFS, we discuss the application of MFS in pan-genome analysis using the maize genome (B73v5) as a multi-omics utility case study. The pan-genome represents the entire set of genes within a species (19), consisting of a ‘core’ genome, containing gene models shared between all individuals of the species, and the ‘non-core’ genome, made up of near-core, dispensable, and private gene models occurring in most, some or a single genome, respectively. Plant genomes are highly dynamic, and several challenges remain to be overcome before cost-effective and rapid pan-genome construction is possible (20). Therefore, we provide modules aimed at tackling problems associated with pan-genome analysis by applying machine learning algorithms and classifying genes as core or non-core in a new genome using only multi-omics data associated with the genes.
Materials and methods
Overview of the maize feature store database
We have created an application that uses a MongoDB database (NoSQL) named ‘BigFeatureDb’. MongoDB is a document-oriented data store that stores data in collections. Collections are made up of documents, and each field in a document is associated with a value. Complex maize omics data has been imported into these embedded data models via the Pymongo library. We stored each omics data type in separate collections for each feature type (e.g. ‘DNASequenceFeatures’). These collections contain documents corresponding to the gene model set of the B73v5 reference genome (21). The document’s key is used as the MongoDB primary key. Within each document, field-value pairs are used to hold pairs of gene model feature names and feature values. This database structuring allows a variety of aggregation operations to process complex queries.
Maize feature store architecture
The Maize Feature Store has three layers, transform (to ingest and process data and create features), store (for storing the created features and their metadata), and serve (to make available the stored features). The data in the Maize Feature Store is stored in the MongoDB database, and the features are extracted and pre-processed from varied sources using customized Python scripts. The front-end application in the Python Flask framework makes the data available to various end-users.
Application development
We developed an interactive web-based query system to retrieve the desired information from the maize reference genome version B73v5 omics data using Flask, HTML5, JavaScript and CSS. The server-side scripting uses Python code and Pymongo (v3.11.3) drivers. A sophisticated search query system enables users to conduct multiple searches, data visualization and modeling.
The graphical user interface is designed to help users conduct an automatic end-to-end analysis of the maize omics data, along with basic exploratory analysis and predictive modeling of the datasets. To do this, the interface is divided into sections and subsections in the form of various menus on the navigation bar. The home page (https://mfs.maizegdb.org/) illustrates the overall functioning of the tool with three major components (‘Features and Analysis’, ‘Models’ and ‘More’) for getting started with the analyses.
The ‘Features and Analysis’ module (https://mfs.maizegdb.org/features) is divided into three main sections: All data analysis, Downsampled analysis, and User candidate gene analysis. Each of these sections is further subdivided into Sequence Features, Gene Structure Features, Expression Features, Chromatin Features, Count Features, Correlation Features and Other Features. These sections have additional subsections with specialized functions that operate dynamically on the selected dataset. Users can select their desired features and labels in each subsection and carry out a wide range of analyses using tables and graphs. Each subsection can analyze either the entire dataset or a randomly down-sampled dataset. The outputs of the selected analysis (tables and graphs) are displayed reactively on a separate webpage. The user can download all the tables (copy or .csv or .xlsx or .pdf) and plots (.png) using specified buttons. Additionally, tables and graphs are interactive, allowing for deeper data exploration. It is crucial to note that some subsections, such as ‘DNA Sequence’ Features, do not display the whole dataset to prevent the complexity of selecting hundreds of features and avoid the visualization becoming unwieldy. However, users can always download the selected subset or the complete dataset via the ‘Download Source’ or ‘Download All’ choices. All the front-end structures were created using Bootstrap (v4.0), jQuery (v3.5.1), and Flask (v1.1.2) Python packages. The plots were built by Dashbio v0.7.1 and plotly (v5.3.1)/matplotlib (v3.4.2), respectively.
The ‘Predictions’ section consists of machine-learning models as a web service. As a use-case, we provide two models: the ‘Advanced’ model (https://mfs.maizegdb.org/model_advanced) and the ‘Basic’ model (https://mfs.maizegdb.org/model_basic), for classifying maize core and non-core genes (21). Two simple forms are built using HTML and CSS to take input from the users on the top 25 features that were highly predictive for differentiating between core and non-core genes. Our application uses a Gradient Boosting Classifier for the ‘Advanced’ model and a Random Forest Classifier for the ‘Basic’ model, both built with scikit-learn (v1.0.2) and wrapped in Flask.
The ‘More’ section holds additional information for the smooth functioning of the interface, such as links to the Data Sources, Tool Sources, Frequently Asked Questions and Contact Page.
Data acquisition
The central idea behind generating and extracting a broad set of omics data associated with the maize genome is to allow researchers to explore these intrinsic and extrinsic gene features and conclude their research findings linked to any eukaryotic organisms or, more specifically, to maize.
We curated an extensive set of genomics, transcriptomics, epigenomic, variomic and proteomics data from three major sources: MaizeGDB, peer-reviewed publications and data generated in other labs (https://mfs.maizegdb.org/data_sources). The B73v5 maize gene models, canonical protein sequences and coding sequences were collected from the MaizeGDB database. Gene structural features were extracted from the annotation files (GFF) linked to the B73v5 genome. The gene expression (mRNA and protein abundance) datasets across multiple tissue types and conditions were collected from peer-reviewed publications and from other labs. The epigenomic and variomic datasets were gathered from MaizeGDB JBrowse (6) and the maize Nested Association Mapping paper (21).
Sequence feature generation
We used the canonical transcript and protein sequences to generate the sequence features for genes with multiple transcripts. The coding sequence data were used for generating various numerical representation schemes of DNA sequences. Four modules of the rDNAse package (22), basic tools, nucleic acid composition, autocorrelation and pseudo nucleotide composition (details on the DNA features can be found here: https://mfs.maizegdb.org/DNAseq) were used to generate DNA sequence features. The genomic sequences were also used to generate various codon and amino acid usage features such as the codon adaptation index, expected effective number of codon and stacking energy using the SADEG package (23).
Numerous structural and physicochemical descriptors, such as amino acid composition, autocorrelation, composition/transition/distribution (CTD), conjoint triad, quasi-sequence order, pseudo amino acid composition and the amphiphilic pseudo-amino acid composition (details on the protein sequence features can be found here: https://mfs.maizegdb.org/Proteinseq), were extracted from the peptide/protein sequences using the protr package (24). The protein sequences were also used to generate predicted protein subcellular localization features (nucleus, cytoplasm, extracellular, mitochondria, cell membrane, endoplasmic reticulum, plastid, golgi apparatus, lysosome/vacuole, peroxisome) using the WolfPsort (25) and Deeploc (26) programs The protein structural features such as coils, hot loops, transmembrane helices and signal peptides were predicted from the amino acid sequences as an input using DisEMBL (27), TMHMM (28) and SignalP (29), respectively.
Structure feature generation
The gene annotation (GFF) files linked to the B73v5 maize genome were used to extract numerous gene structural features such as the gene length, number of isoforms, exon length, average exon length, number of exons, chromosome associated with each gene, coding sequence length, five-prime untranslated regions (UTR) length and three-prime UTR length using customized Python script. The Python script parses through the GFF file to generate these features.
Distance features such as distance from the chromosome center, distance to the nearest knob, the centromere and the telomere were also generated for each gene of the B73v5 maize genome. The data were downloaded from MaizeGDB.
Expression feature collection
The maize transcriptomics and proteomics data consist of expression levels for each gene across multiple tissue types and experimental conditions. The RNA expression features included data from the MaizeGDB qTeller (30) B73v5 instance. The MaizeGDB qTeller contains almost 200 unique datasets from 12 projects. Each dataset was mapped with a consistent pipeline to provide fair comparisons. Any future datasets added to the MFS will follow the same pipeline. The B73v5 instance of qTeller contains data from eight studies from multiple labs (31–38) covering 172 tissues/conditions. The ‘Compare RNA & Protein’ tool of qTeller incorporates data from a single mRNA and protein study (33) spanning 23 tissues/conditions. Apart from gene expression, we estimated the average mRNA abundance level, protein abundance level, maximum mRNA abundance level, maximum protein abundance level, tissue gene abundance breadth and tissue protein abundance breadth for each gene across all tissues and conditions. The breadth is defined as the number of tissues where the gene or protein showed expression.
Chromatin feature generation
Chromatin features comprised of chromatin states, three histone modifications (H3K4me3, H3K27me3, H3K27ac), open chromatin as quantified by ATAC-Seq and DNA methylation (quantified separately in CG, CHG and CHH contexts) were obtained from the ChromHMM software and Dai, Xiuru et al. (1). The chromatin states were generated from ChIP-Seq data (including nine types of histone modifications, H2AZ, H3, H3K4me1, H3K4me3, H3K9ac, H3K27ac, H3K27me3, H3K36me3, H3K56ac) in two tissues, ear and leaf (39). Histone modifications are often found in recurring combinations at promoters, enhancers and repressed regions. These combinations are called ‘chromatin states’ and can annotate regulatory regions in genomes. We have included multiple chromatin states features from ChIP-Seq data using the tool ChromHMM (A multivariate HMM for chromatin combinatorics) (40).
Count feature generation
We generated the ‘Count’ features by finding and counting annotations from multiple genome interval files whose genomic coordinates overlapped with the maize gene sites using the bedtools suite (41). The genome annotation files included the MaizeGDB B73v5 JBrowse annotations (mutational insertions, transcription factor binding sites, transcription start sites, enhancers, transposable elements, miRNAs) (39, 42–47) and G-quadruplexes. The G-quadruplex annotation files were generated using in-house Python scripts from the B73v5 maize genome sequence. Counts were computed for three genomic regions: the first region included the gene body, the second included a 1KB region upstream and downstream of the gene start and end sites and the third covered a much larger region, comprising 5 KB upstream and downstream of the gene start and end site.
Correlation feature collection
The correlation features include 12 co-expression modules identified through weighted gene co-expression network analysis. The data comprise 79 tissues, six-organ developmental gene atlas coupled with five abiotic/biotic stress transcriptome datasets (48). These topology features were available for B73 AGPv4 gene models; therefore, B73 AGPv4 gene models were converted to B73v5 using a conversion list published on MaizeGDB.
Varionomic feature generation
Varionomic features included the count of single nucleotide polymorphisms (SNPs) per gene model and the effects of SNPs on the genes. The count of SNPs per gene model was calculated by finding overlapping regions between the SNP data VCF file from (21) and maize gene coordinates using Bedtools, and SnpEff (49) was used to annotate and predict the impact of variations on genes. This tool takes pre-defined variations listed in a VCF file containing the nucleotide change and its location and predicts if the variants are detrimental.
Other feature generation
The ‘Other’ feature section includes evolutionary gene age (described below) and the total number of presence/absence of associated Pfam-domains per gene model (21, 50). The direction and magnitude of natural selection were inferred from the ratio of nonsynonymous substitutions (Kn)/synonymous substitutions (Ks) between Sorghum and maize B73v5 orthologous genes and from the ratio of nonsynonymous substitutions (Kn)/synonymous substitutions (Ks) between maize Tzi8, a tropical maize line (21), and maize B73v5 orthologous genes. Ks and Kn values were derived between syntenic ortholog coding sequences of B73v5 and Sorghum bicolor v3 (https://phytozome-next.jgi.doe.gov/info/Sbicolor_v3_1_1) using the tool CoGe SynMap (51) (https://genomevolution.org/coge/SynMap.pl) with the parameters Relative Gene Order; -D 20; -A 5; Quota Align Merge; Syntenic Depth B73:Sorghum 2:1; and CodeML Kn/Ks. Ks and Kn values between B73v5 and the maize tropical cultivar Tzi8 were derived using similar parameters except the Syntenic Depth was set to 1:1.
The evolutionary gene age was calculated by searching for homologs within increasingly broad clades using the phylostratr pipeline (52). The deepest clade that contains a homolog of the protein(s) encoded by a gene is that gene’s age as described by Arendsee, Zebulun et al. (52). The maize gene age is classified into 21 categories based on the presence/absence of the homologs of maize genes in 20 representative eukaryotic species (including cellular organisms, Andropogoneae, commelinids, Embryophyta, Eukaryota, Liliopsida, Magnoliopsida, Mesangiospermae, PACMAD clade, Panicoideae, Petrosaviidae, Poaceae, Poales, Spermatophyta, Streptophyta, Streptophytina, Tracheophyta, Tripsacinae, and Viridiplantae).
Label generation
In addition to the different genomics, proteomics and transcriptomics features, the Maize Feature Store also includes example biological annotations. They can be used as class labels for users looking to classify their genes of interest to any of the biological annotations or identify relationships between these gene annotations and a variety of features offered through the MFS. These gene annotations are not only meant to act as targets, but are also intended to function as features when appropriate. For example, we can use whole-genome duplication (WGD)/tandem gene annotations as features when trying to solve core/non-core gene prediction problems and vice versa. Currently, MFS contains three sample labels: ‘Classical’ (classical/other) genes, ‘Pan-genome’ (core/near-core/dispensable/private) genes, ‘Gene Origin’ (WGD/tandem/both) genes, and a ‘No Label’ option. Classical genes are the most well-studied genes in maize, most of which have a visible mutant phenotype (for example, liguleless2) as described by Schnable, C, James et al. (53). We downloaded 430 maize classical genes from MaizeGDB (Classical Genes). The core/near-core/dispensable/private genes and WGD/tandem/both genes were collected from maize pan-genome generated as part of the Nested Association Mapping (NAM) genome sequencing project (21). The ‘No Label’ option lets users view the relationship between the genes independently of any annotations. This selection is provided to enable users to view the properties of all genes without labeling them into different gene categories or annotations. Using this feature, users can examine the features of multiple genes and can choose to annotate them based on common patterns identified between different genes. As it involves the inspection of all the genes, they work only for the “Submit for analysis” button.
Data visualization
The MFS user interface is pre-configured with plotly, matplotlib, and Dashbio allowing innovative visualizations such as data distributions, connections between features, and aggregate statistics (minimum, maximum, average, unique categories, outliers, missing values, etc.). This enables researchers to gain rapid insight into the features and make more informed decisions about using specific features. The interface also provides detailed instructions on the usage and interpretation of each plot. Users are given options to conduct each exploratory analysis using the entire omics dataset or the downsampled data using the ‘Submit analysis’ and ‘Downsampled analysis’ buttons.
Downsampled analysis
The ratio of label categories is frequently uneven, resulting in a bias favoring the majority class. For example, seventy-two percent of our genes are marked as core in the maize reference genome version B73v5, and twenty-eight percent are annotated as non-core (near-core, dispensable and private genes). Therefore, we offer the random down-sampling method to address the issue of unbalanced data during exploratory analysis and provide users with the option of ‘Downsampled analysis’. It is important to note that the size of the downsampled data is different for each label (Classical/Pan-genome/Gene-Origin) selection as the size of the minority class is different in each label.
User candidate gene analysis
The user candidate gene analysis section allows users to do a comparative study on their genes of interest. Users can enter a single gene of interest or a group of candidate genes linked to specific biological pathways or functions and compare them with other down sampled sets of maize genes. Two types of analyses are possible for the user-defined candidate genes: a) single candidate gene analysis and b) analysis of multiple candidate genes. For single gene analysis, users can enter a single gene of interest and visualize the output for the selected features either in tabular format or graphical format with a marginal plot showing the frequency distribution of the selected gene features for all maize genes along with highlighting the candidate gene (Supplementary Figure S1A). For analysis of multiple candidate genes, users can enter a list of genes and compare their gene list for the selected feature with the other downsampled maize genes in various univariate or multivariate plots. When using multiple candidate genes, it is recommended that a larger gene list be entered (fifty or more) so that a more reliable comparison of the candidate genes and the downsampled other maize genes can be made. The down-sampling is random based on the number of candidate genes, therefore a larger candidate gene list requires more down sampled genes, resulting in a better representation of the population.
To demonstrate the potential use case of the user candidate gene analysis, we gathered a set of fifty stress genes differentially expressed between the control and salt stress samples (54) and used them to identify unique characteristics common among salt stress genes (Supplementary Figure S1B). Using our univariate analysis, we found that the maize B73v5 salt stress genes differed significantly from other downsampled maize genes regarding the gene structural features of isoform count, coding sequence length, three-prime UTR length, and five-prime UTR length. These structural features showed a significantly higher range among the candidate genes. Previous work on stress genes has also discovered that 3ʹUTR-based mRNA stability controls are present in stressed cells (55), thereby further supporting our findings from the salt stress genes.
Exploratory analysis
The exploratory analysis module in the Maize Feature Store assists users in visualizing all accessible features and labels in tabular and graphical formats after initial preprocessing, cleaning, and normalization steps. Omics datasets come in diverse scales and follow their own statistical distributions as they are collected from disparate sources; therefore, data standardization becomes crucial for omics datasets. The MFS application allows for the normalization of omics numerical features by centering the features with their mean and the standard deviation between 0 and 1 using the StandardScalar function of Sklearn.
Apart from providing fundamental functionality, high-end modules in MFS calculate and perform various univariate, bivariate, or multivariate analyses such as Histograms, Count and Distribution plots, Pair plots, Box plots, Violin plots, Joint plots, Scatter plots, Correlation plots, Categorical Bar plots, Heatmaps, Clustering plots, and Dimension reductions (PCA) (see Supplementary Methods). However, the ‘Gene Expression’ dataset currently provides a preview of the results by limiting the display of Histograms, Count and Distribution plots, Pair plots, Box plots, Violin plots, Correlation plots, and Heatmaps to five tissues of the selected lab. Since each lab includes multiple tissues, the limit of visualizing five tissues is intended for better analysis and visualization of plots. Users can modify the script to view more than five tissues from a lab. Most of these plots have options to download, zoom-out/zoom in, reset axes, autoscale, toggle spike lines, show the closest data on hover, compare data on hover, box select, pan, and lasso. Users can also select specific legends to view data only for the selected legends. The Histograms, Count and Distribution plots, and the Categorical Bar plots also come with a two-sided p-value analysis displayed at the top of the selected feature chart to determine whether there is enough statistical evidence in favor of a hypothesis (there is a difference in the selected feature values or frequencies across the different categories of the target variable). For comparing the effect of the selected continuous feature on the classical/other genes target variable (binary), we carry out a two-sample test using the scipy.stats library in Python. For comparing the effect of the selected continuous feature across multiple categories of the target variable such as core/near-core/dispensable/private genes or WGD/tandem/both genes, we carried out a one-way ANOVA test using the Python stats library, and lastly, for comparing the effect of the selected categorical feature across two or more categories of the target variable, we carried out the Chi-square test using the scipy.stats library in Python.
Details on the usage and interpretation of all the plots and tables are also available on the MFS website (https://mfs.maizegdb.org/Structure) and Supplementary Methods. While MFS is intended to facilitate plot generation using a graphical user interface, by hiding sophisticated plotting routines behind MFS modules, users can download the appropriate module Python script for direct replication and transformation of the visualizations.
Data clustering
The MFS uses advanced functionalities to analyze unlabeled omics data rather than labeled data to overcome the lack of manual annotations. The module can efficiently compute several unsupervised clustering algorithms on downsampled omics data and provides interactive visualization of the results using Dendrograms, Heatmaps, Hierarchical Scatter plots, Hierarchical Heatmaps, and PCA plots (2D, 3D, biplot) (see Supplementary Methods). Different user options are available for some of these modules to dynamically show different results. For example, in the Hierarchical Scatter plots, the ‘Choose Clusters’ option is available where the users can manually enter the number of clusters to visualize in the Pair plot. However, it is recommended that users enter the number of clusters as per the output generated by the Dendrogram plot. To save time and complexity, we limited the Heatmap plot to only display the relationship between the first hundred down-sampled genes and the selected attributes; however, users with sufficient resources are free to utilize the function and customize it to include as many genes as necessary for their specific analysis.
Results
Maize feature store workflow
Maize omics data are generally large, complex, and contain a variety of structures. The ability to store and retrieve data effectively is critical in maize research. Historically, huge datasets have been kept as flat files on disk or relational databases. These platforms are difficult to develop, maintain, and adapt to big-data applications because they adhere to inflexible table structures and frequently lack scalability, such as data aggregation. Therefore, we proposed to design our application to carry out complex operations, including 1) Flexible, to handle a wide variety of data types. This enables researchers to rapidly evolve data models and conduct customized analyses. 2) Scalable, permitting researchers to easily explore large and complex datasets without waiting long periods for simple queries. 3) Operationally mature, including end-to-end encryption, fine-grained data access control, and operational tooling. These operations can facilitate the management of multi-omics data and the accurate alignment of genes across multiple datasets, thereby increasing the feasibility of multi-omics integrative analysis.
Numerous biological prediction problems (56) are based on standard feature sets such as gene length, exon number, and gene expression. These conventional feature sets are repeatedly utilized to tackle many different biological problems and to obtain these features from raw data requires users to know bioinformatics, such as annotating gene models from a genomic fasta file, mapping RNA-Seq reads to genomes or extracting counts of exons per gene model. These processes become tedious and repetitive if we use the same features to solve further biological problems A feature store allows researchers to overcome this obstacle and improve the usability of the omics in the genotype-to-phenotype context. We developed the Maize Feature Store tool to simplify the management, access, and analysis of omics datasets for a wider range of users.
Application of MFS on pan-genome classification
We illustrate the capability of the Maize Feature Store in applying and analyzing multi-omics data for classifying genes as core or non-core and identifying top omics features that are most helpful in predicting their classification within a pan-genome. As reported in Figure 1, several modules were developed to follow a precise exploratory analysis workflow that goes from the data selection to the downstream data analysis and ultimately to modeling. For our case study, we developed two models: one that utilized all omics features (a total of 14 407 features) (https://mfs.maizegdb.org/feature_details) and another that utilized a subset of omics features (a total of 10 271 features) consisting of only the gene structure, gene sequence, and protein sequence data (https://mfs.maizegdb.org/feature_details). The model development lifecycle involved several stages, such as feature engineering, dealing with imbalanced data, feature selection, model building, hyperparameter tuning, and finally selecting the most optimal model (see Supplemental Methods).
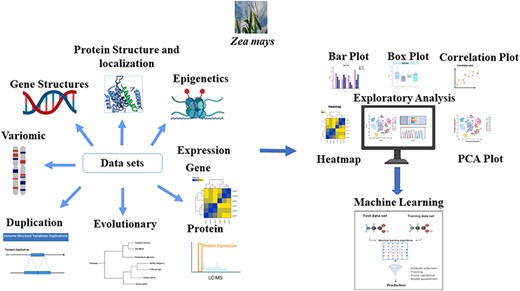
Module description: The MFS consists of three main modules: Features, Downstream Analysis, and Modeling. We assembled the omics features associated with each gene model in Zea mays (B73v5) based on various sources as indicated by the ‘Data sets’ arrows of the figure. Many preliminary and advanced exploratory analyses can be performed on the generated features as indicated by the ‘Exploratory analysis’ module of the figure. Systematic evaluation of machine learning (ML) approaches is used in the Modeling section to solve complex biological problems, such as pan-genome prediction. The Graphical Overview was created using BioRender.com.
An example of the ‘Data Table’ module is shown in Table 1. In the ‘Data Table’ module, it is possible to view all the genes and the selected features. Users can sort the table columns and use the search bar to look up specific gene IDs. We used the MFS data exploration and visualization modules to perform several univariate, bivariate, and multivariate analyses of the core and non-core gene structural features (Supplementary Figures S2-S6, see Supplemental Methods). An initial analysis of the data provided a quick visual summary of the potential association between the selected features of interest and the various categories of the ‘Pan-genome’ label (Figure 2 and Supplementary Figures S2-S6). By simultaneously exploring gene structure features, we can observe that several features are significantly correlated in both core and non-core genes. Therefore, the plots can initially demonstrate how the different genomic features can contribute to our understanding of core or non-core genes and highlight the potential for gene structural features in pan-genome classification.
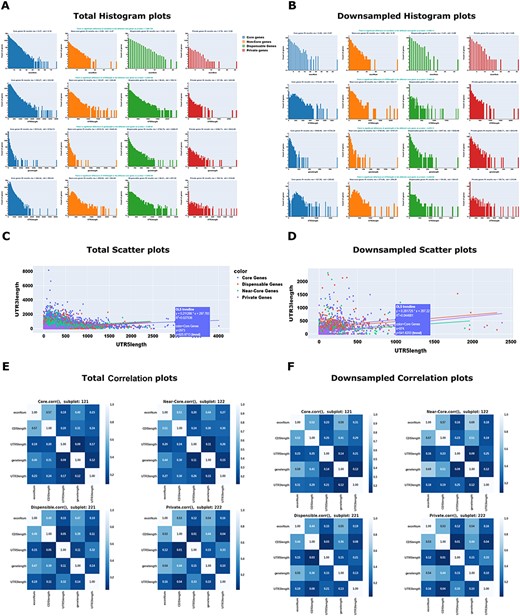
Example Maize Feature Store outputs. The MFS provides users with options to carry out several univariate, bivariate, and multivariate analyses for both the total and downsampled omics data. Univariate analysis example: (A) Total Histogram; (B) Downsampled Histogram; Bivariate analysis example: (C) Total Scatter plot; (D) Downsampled Scatter plot; Multivariate analysis example: (E) Total Correlation plot; (F) Downsampled Correlation plot. These plots were generated from the selected Gene Structures such as Gene length, Exon number, three-prime UTR length, five-prime UTR length, and the selected label (‘Pan-genome’: core/near-core/dispensable/private). The plot’s colors and legends indicate the multiple ‘Pan-genome’ categories. In addition to the graph, to increase the interpretability of the data, we have also included p-values, mean and standard deviations of the selected datasets. For details on the interpretation of the plots, see (https://mfs.maizegdb.org/Structure).
Dynamic visualization of the selected gene structure datasets (exon number, five-prime UTR length, gene length, three-prime UTR length and the ‘Pan-genome’ categories) using the MFS’s ‘Data Table’ option. Only ten rows are displayed per page
| ID | ExonNum | UTR5length (base pairs) | Genelength (base pairs) | UTR3length (base pairs) | PanGenome_label |
|---|---|---|---|---|---|
| Zm00001eb000010 | 9 | 105 | 5588 | 1668 | Near-Core Gene |
| Zm00001eb000020 | 9 | 849 | 5549 | 313 | Core Gene |
| Zm00001eb000050 | 7 | 645 | 5829 | 0 | Dispensable Gene |
| Zm00001eb000060 | 2 | 299 | 1023 | 364 | Dispensable Gene |
| Zm00001eb000070 | 6 | 0 | 8641 | 0 | Dispensable Gene |
| Zm00001eb000080 | 9 | 447 | 3132 | 730 | Near-Core Gene |
| Zm00001eb000100 | 6 | 82 | 3105 | 641 | Near-Core Gene |
| Zm00001eb000110 | 2 | 15 | 821 | 43 | Dispensable Gene |
| Zm00001eb000120 | 1 | 0 | 628 | 268 | Near-Core Gene |
Unified features excel over individual subsets in maize gene classification: core vs. non-core categories
The “Modeling“module of MFS offers an “Advanced Model” form and a”Basic Model” form which allows users to make predictions for their genes based on certain inputs. We trained the “Advanced’’ model using the top 25 features from a comprehensive set of omics features generated using a Hybrid Feature Selection method and a base Gradient Boosting Classifier with five-fold cross-validation (Supplementary Tables S1-S2 and Figure 3A). We built a simplified “Basic” model by training on the top 25 features generated using a similar approach from only the gene structural features and sequence features (Supplementary Tables S3-S4 and Figure 3B). To evaluate the specific contributions of each feature type to the overall accuracy of core and non-core gene prediction, we performed individual predictions using the other distinct subsets of features (Expression Features, Chromatin Features, Count Features, Correlation Features, and Other). This involved constructing separate machine-learning models for each feature subset (Supplementary Figure S11-S16). We tested the performance of six machine-learning algorithms for the classification of “Pan-genome” genes on both “Advanced’’ and “Basic” models, namely: (1. Logistic Regression, 2. Random Forest Classifier, 3. Gradient Boosting Classifier, 4. Extra Trees Classifier, 5. KNeighborsClassifier, and 6. SVM Classifier) and two distinct optimization approaches (1. Random and 2. Grid Search). In general, all five approaches performed well, but Gradient Boosting Classifier performed significantly better in the “Advanced” model with the area under the Receiver Operating Characteristic Curve (AUC-ROC) = 0.85, Average Precision-Recall (PR) = 0.96, and F1 = 0.92 (Figure 3C) and the Random Forest Classifier performed significantly better in the “Basic” model yielding an AUC-ROC = 0.80, Average PR = 0.92 and F1 = 0.89 (Figure 3D). We compared the results to random classification to gain a proper perspective on the model performances. Based on random classification, the AUC-ROC would be 0.5. When AUC = 0.5, the classifier cannot distinguish between positive (core) and negative (non-core) class points, as the classifier is predicting a random class or a constant class for all the data points. An increase in AUC-ROC and F1 can be seen in the “Advanced” model, especially compared to the “Basic” model. Therefore, our performance increased significantly when we used both intrinsic and extrinsic features, as demonstrated by the “Advanced” model.
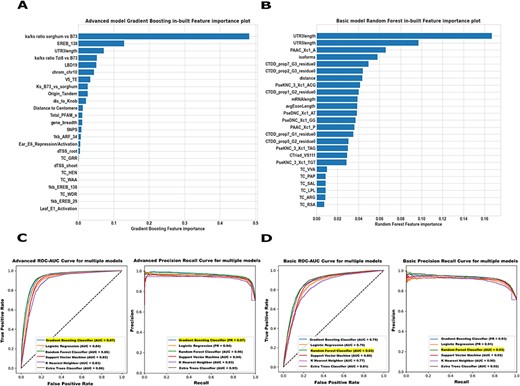
Maize Feature Store example Basic and Advanced models. (A) In our Advanced model, both intrinsic and extrinsic features contributed substantially to the core/non-core gene predictions in maize B73v5. The 25 omics features were ranked based on how useful the model found each feature in predicting the target (core/non-core genes). (B) The Basic model feature importance plot displays only the structural and sequence features most predictive of identifying the core and non-core genes in B73v5. Higher scores indicate that a specific feature has a larger impact on the model used to predict a specific variable (core/non-core). (C, D) The prediction performance of both the ‘Advanced’ model and the ‘Basic’ model was evaluated across all classifiers on the test set using AUC-ROC (left) and the area under the Precision-Recall Curve AUC-PR (right) metrics. For detailed model evaluation and performance analysis, see the Supplementary Figure S17-S18.
Additionally, both of our models: ‘Basic’ and ‘Advanced’, outperformed a previous model in terms of accuracy recently published by Yocca, E, Alan et al. (57), which predicted core genes of Oryza sativa and Brachypodium distachyon using only intrinsic features such as gene sequence features, evolutionary features, and gene structural features. They achieved an AUC-ROC of approximately 0.77 and an accuracy of approximately 0.71 when trained and tested with the Oryza sativa balanced datasets and an AUC-ROC of approximately 0.86 and an accuracy of approximately 0.80 when trained and tested with the Brachypodium distachyon balanced datasets using a Random Forest method for ML, whereas our ‘Basic’ model (Random Forest Classifier) achieved an AUC-ROC of approximately 0.80 and an accuracy of approximately 0.84 in the testing set, and our ‘Advanced’ model (Gradient Boosting Classifier) achieved an even higher accuracy of approximately 0.89 and AUC-ROC of approximately 0.85. In this way, our models not only classify genes as core or non-core but also challenge the efficacy of current pipelines by comparing model output with pipeline output. Analyses of complex genomes by pan-genome pipelines often result in the incorrect annotation of genes as core or non-core. Our model can provide extra validation to the pipeline output and identify mis-annotations that may occur in the current pipelines, which are both time-consuming and computationally expensive.
Investigating the features that have strong differentiation powers in both the ‘Basic’ and ‘Advanced’ models.
The best performing model (Gradient Boosting Classifier in the ‘Advanced’ model and Random Forest Classifier in the ‘Basic’ model) was used to determine which predictor variables are most significant for prediction performance. In this way, we can gain insights into the biology of core and non-core genes. The 25 most important variables (Figures 3A and 3B) for training the “Advanced‘’ model and the ‘Basic’ model were generated using a Hybrid Feature Selection method and a base Gradient Boosting Classifier as described in the materials and methods section. A Gradient Boosting Classifier has a built-in variable importance assessment. The Kn/Ks ratio of both sorghum vs. B73 and Tzi8 vs. B73, a measure of evolutionary pressures on protein-coding regions, was among the top five most significant features in the “Advanced’’ model. There have been previous pan-genome studies that compared synonymous (Ks) and nonsynonymous substitution (Kn) rates (58). These studies have indicated that dispensable genes undergo more non-synonymous substitutions, as well as increasing Kn/Ks ratios, implying greater positive selection on dispensable genes (59–61). While performing exploratory analysis with the genes, we also observed a difference in the Kn/Ks ratio of Tzi8 vs. B73 among the ‘Pan-genome’ genes with a mean value of 13.90 in the dispensable genes and 4.58 and 4.70 in the near core and core genes, respectively, aligning with results found in the previous studies of greater positive selection on dispensable genes. The two-sided p-value analysis also indicated a significant difference in the Kn/Ks ratio observed among the ‘Pan-genome’ genes. Other important predictors in our ‘Advanced’ model were the difference in the ratio of the WGD regions among the core or non-core genes, presence, and absence of Pfam domains (protein families, domains, and functional sites extracted from the Pfam database) for coding genes in the core genome set and those in the dispensable genome, Transcription Factor Ethylene Responsive Element Binding Factor domain EREB (stress-responsive transcription factors) and (TE) transposable elements.
Gene duplications play a major role in the evolution of novel traits in eukaryotes (62, 63). The WGD regions are found to contain a higher ratio of core and near-core genes, whereas non-WGD regions (tandem regions) contain a higher ratio of dispensable and private genes (64, 65). Additionally, the exploratory analysis also indicated that in our omics dataset, the non-core genes had a higher tandem repeats ratio than the core genes (Supplementary Figure S7). An enrichment of TEs in the vicinity of dispensable genes was reported in B. distachyon (59) and B. oleracea (66). Our model, as well as our exploratory analysis (https://mfs.maizegdb.org/TE), complements the findings of previous studies on transposable elements and Pfam domains (67), as the maize B73 dispensable genes were also found to be enriched with transposable elements around the 1Kb and 5Kb regions upstream and downstream of the gene start site and end site respectively, and the total Pfam domains were also abundant among the maize B73 core genes compared to the dispensable genes. As the EREB transcription factors are involved in plant hormone responses under stress conditions (68), they are more likely to be enriched among dispensable genes than the core genes, and our study confirms this (https://mfs.maizegdb.org/TFbindingSite).
The top features in our ‘Basic’ model having the most influence in the classification of core or non-core genes are the five-prime UTR length, three-prime UTR length, isoforms count, and sequence features such as Composition Transition Distribution (CTDD), pseudo dinucleotide composition (PseDNC) and many more. Most of these features displayed significant differences between the maize B73 core and non-core genes (https://mfs.maizegdb.org/Structure). Earlier studies have also stated that dispensable genes tend to display common features similar to young genes: short gene length, weak homology, low expression, rapid evolution, and turnover (69), thereby further supporting our findings on the topological properties of core and non-core genes.
Discussion
The growing number of omics datasets from diverse sources have highlighted the importance of evaluating specific models and methods for collecting, managing, and analyzing multi-omics data to better explore the interplay between the multiple cellular, molecular, and phenotypic layers. While several multi-layer data structures are available, there is still a need for end-to-end solutions for storing, exploring, and modeling data. To solve this need, we proposed using MFS as a suitable structure to manage commonly used maize omics features. MFS will benefit bioinformaticians, data scientists, and experimental researchers interested in solving complex biological problems Our tool enables researchers to share and discover features, create more effective machine-learning pipelines, and perform exploratory analyses. It provides users without domain knowledge or modeling experience the ability to identify the most significant factors affecting the target problem. For example, during the exploratory analysis of ‘Pan-genome’ genes (Figure 2), we observed that the exon number varied across the pan-genome categories and thus might be a strong predictor of core or non-core genes.
An example of a current application of these models involves classifying genes in a new species closely related to maize as core or non-core without constructing an expensive pan-genome. Our models outperform random assignment for most downstream applications with around 90% accuracy. Our model would also be ideal for newly sequenced or poorly annotated genomes. Where other tools like BLAST could also infer annotation, it does not provide underlying insights for the assignments beyond sequence homology.
Each year, numerous papers and research articles are published on maize, utilizing omics data. However, although data repositories exist, there is a need to extend model orgaism databases like MaizeGDB to provide end-to-end data analysis. MFS, in this context, provides a central hub of maize omics features with flexible and expandable functionality that enables maize researchers to configure the tool for specific analyses. Additionally, MFS’s modeling module utilizes a comprehensive set of omics features to conduct a core/non-core gene classification. Even though several prediction or classification problems have been addressed using a wide range of omics features in mice (70), D. melanogaster (71, 72), and C. elegans (73), no work on plants, more specifically maize, has been reported. We were able to build a classification model utilizing the comprehensive set of features (‘Advanced’ model) and perform a comparative study by building another model utilizing just sequence and structural features known as the ‘Basic’ model. Although the ‘Basic’ model was more generalized, the ‘Advanced’ model performed significantly better (Figure 3C), thus showing that an elaborate assembly of intrinsic and extrinsic factors from a wide range of sources covering multiple aspects of a gene greatly outperforms the approach based solely on sequence or structural features. We further emphasized the necessity of using both intrinsic and extrinsic features by comparing our models (both ‘Basic’ and ‘Advanced’) with already existing models by Yocca, E, Alan et al. (57), which predicted core and non-core genes of Oryza sativa and Brachypodium distachyon, respectively. Our ‘Advanced’ model performed significantly better with an accuracy of almost 25% higher than their same species Oryza sativa model (trained and tested on the Oryza sativa balanced datasets) and almost 11% higher than their Brachypodium distachyon model (trained and tested on the Brachypodium distachyon balanced datasets).
In this work, we aimed at the needs of both experimental and computational researchers. We addressed the need for resources that bridge the gap between the growing number of omics datasets and their potential as training data for modeling and machine learning. We developed a framework that hosts over 14 000 gene-based machine learning features built on multi-omics data to facilitate the exploration and modeling of classification problems The tool’s modularity will allow computational researchers to add additional functionality, fine-tune existing functionalities, and reproduce the entire application for other species of interest.
Supplementary material
Supplementary material is available at Database online.
Data availability
Project name: Maize Feature Store (MFS); Project home page: MFS is freely available on GitHub at https://github.com/shatabdi123/MFS_Application Web version of MFS is available at https://mfs.maizegdb.org/. The dataset for MFS can also be accessed on Kaggle: https://kaggle.com/datasets/332177dbd2271966f2291640acf6f7057bde915d939b3bf67 545a5f24a0e3fe3. Programming language: Python, R, JavaScript, HTML, CSS; Other requirements: Flask 1.1.2 or higher. The application is platform independent.
Abbreviations
MFS (Maize Feature Store), ML (Machine Learning), WGD (Whole Genome Duplication), SNP (Single Nucleotide Polymorphisms), VCF (Variant Call Format), GFF (General Feature Format), AUC-ROC (Area under the Receiver Operating Characteristic Curve), GUI (Graphical User Interface), AUC- PR (Area under the Precision-Recall Curve)
Funding
This research was supported by the US. Department of Agriculture, Agricultural Research Service, Project Number [5030–21000-068-00-D] through the Corn Insects and Crop Genetics Research Unit in Ames, Iowa. This material is based upon work supported by the Department of Agriculture, Agricultural Research Service under Agreement No. 58–5030-0-036 [Iowa State Award: 022172–00001 to J.W.W.]. Mention of trade names or commercial products in this publication is solely for the purpose of providing specific information and does not imply recommendation or endorsement by the U.S. Department of Agriculture. USDA is an equal opportunity provider and Employer.
Conflict of interest
None declared.
Acknowledgements
We thank the research groups of the Iowa State University and USDA-ARS, Corn Insects and Crop Genetics Research Unit and Dr Rita Hayford for their constructive feedback, which has contributed to the improvement of our platform.

{kind=link}
{kind=link}
{kind=link}