





Abstract
Curated resources at centralized repositories provide high-value service to users by enhancing data veracity. Curation, however, comes with a cost, as it requires dedicated time and effort from personnel with deep domain knowledge. In this paper, we investigate the performance of a large language model (LLM), specifically generative pre-trained transformer (GPT)-3.5 and GPT-4, in extracting and presenting data against a human curator. In order to accomplish this task, we used a small set of journal articles on wheat and barley genetics, focusing on traits, such as salinity tolerance and disease resistance, which are becoming more important. The 36 papers were then curated by a professional curator for the GrainGenes database (https://wheat.pw.usda.gov). In parallel, we developed a GPT-based retrieval-augmented generation question-answering system and compared how GPT performed in answering questions about traits and quantitative trait loci (QTLs). Our findings show that on average GPT-4 correctly categorized manuscripts 97% of the time, correctly extracted 80% of traits, and 61% of marker–trait associations. Furthermore, we assessed the ability of a GPT-based DataFrame agent to filter and summarize curated wheat genetics data, showing the potential of human and computational curators working side-by-side. In one case study, our findings show that GPT-4 was able to retrieve up to 91% of disease related, human-curated QTLs across the whole genome, and up to 96% across a specific genomic region through prompt engineering. Also, we observed that across most tasks, GPT-4 consistently outperformed GPT-3.5 while generating less hallucinations, suggesting that improvements in LLM models will make generative artificial intelligence a much more accurate companion for curators in extracting information from scientific literature. Despite their limitations, LLMs demonstrated a potential to extract and present information to curators and users of biological databases, as long as users are aware of potential inaccuracies and the possibility of incomplete information extraction.
Introduction
The value of biological databases comes from efforts from dedicated and knowledgeable personnel who gather, manage, and display curated data in a centralized location [1]. Curators are often trained PhD-level professionals who possess domain knowledge and spend a substantial amount of time integrating unstructured or non-standardized structured data provided in journal articles and supplementary materials. While standardized data reporting and curation processes could lead to significantly improved automation and database integration, in current literature, data are highly heterogeneous, non-standardized, or scattered, requiring a large degree of manual curation. Often, the curation process requires verification of data and correction of minor mistakes like typos prior to uploading the curated data into data management systems. Within database projects, curators ensure that the datasets are formatted correctly and that identifiers follow community data standards [1, 2]. Because of the long training and required dedication, good curators are hard to find, especially for biological databases.
The standardization of published data has long been the goal of groups like The International Society for Biocuration (https://www.biocuration.org). However, until publishers agree and insist on data in common formats upon article submission, curators must use software tools like Excel and Emacs to transform the data in tables into uploadable files that fit the database schema. An important step in curation is identifying relevant literature that would most enrich the database, with an effort to group manuscripts based on general categories [2]. The next curatorial step involves capturing crucial research outcomes from the selected manuscripts and their incorporation into the database. This process ensures the long-term accessibility to diverse genetic mapping association results, facilitating the selection of breeding goals, and improving the understanding of the genetic basis for diverse and complex traits [2]. Careful curation considers the inconsistency in the data structure, as the same quantitative trait loci (QTLs) can be listed as a named QTL in a table within a paper or simply as marker–trait association (MTA) in the supplemental data of another. Rather than uploading all data as a “stand-alone” piece of work, care to create links and synonyms creates relationships among projects and can illustrate the progress, e.g. from a QTL to candidate gene for a given trait.
Large language models (LLMs) are based on training on words and relationships between words using statistics and machine learning methods. They are part of a suite of natural language processing approaches that goes back to the 1950s [3]. In the past decade, natural language processing got a big boost through the use of deep learning methods, such as Word2vec [4]. LLMs have been successfully used in various biological and nonbiological applications, scientific writing of journal articles and grants [5], agriculture and plant sciences [6–8], and biomedicine [9–11]. Existing LLMs can be augmented by providing instructions to retrieve information from domain-specific tools and databases through Web API calls [12, 13]. While LLMs are pretrained on a large corpus of text, their use can be extended to unseen and user-provided documents using retrieval-augmented generation (RAG), without which LLMs are limited to data that existed during the training process [14–16]. The benefit the RAG framework introduced is the ability to extract relevant information from text, such as manuscripts, by searching for similarities between the user-provided prompt and specific segments from the custom document, providing a more informed response [15].
Given the limited funding and scarcity of personnel in biocuration, we wanted to test whether using LLMs, such as GPT (GPT stands for generative pre-trained transformer; https://platform.openai.com/) [17, 18], can assist with and reduce curatorial workload or increase curated data content independently of manual curation. Publicly available biocuration tools, such as Pubtator [19], and the more recent, artificial intelligence (AI)-powered, PubTator 3.0 [20], are powerful tools for extracting relevant biological information, such as relationships between genes, diseases, and chemicals, from manuscripts [9, 21–24]. Despite the continuous improvements of LLM-enabled biocuration methods, LLMs remain prone to generating incomplete, incorrect, and misleading responses [25, 26]. Until the performance of LLMs achieves parity with the precision of manual curation, curators and users of biological databases should exercise caution in respect to LLM-generated biocuration results and continue to rely on traditional manual biocuration methods [25]. The question we were after was whether generative AI systems, specifically GPT, can perform at the level of a well-trained human curator. It is important to note that we are not looking for a way to replace curators because there is still a strong demand for domain experts in biocuration, but whether GPT can provide a useful level of assistance to curators and users of biological databases, by facilitating information extraction [27]. The capabilities of generative AI applications are rapidly advancing, raising important considerations for the future of humanity [28]. Pretrained LLM models, such as GPT, demonstrated that they have the potential to extract information and present it in a humanlike style that increases comprehension [29].
In this paper, we aim to test the performance and limitations of using generative AI LLM methods when working with different aspects of genetic mapping results obtained from manuscripts. To benchmark our results, we used a representative sample of 36 wheat and barley genetic mapping peer-reviewed journal articles that were manually curated by a professional PhD-level biologist and submitted to the GrainGenes database [30]. Due to the lack of standard genetic mapping curation benchmarks, performance was manually assessed. Furthermore, LLM “hallucinations,” i.e. wrong answers provided with equal confidence as the right answers, previously described from crowd-sourced testing of GPT [26], are discussed in the context of genetic mapping data extraction. We consider how LLMs can be used to assist both the curator and the users of biological databases. For the curators, LLMs can be used to rapidly scan manuscripts and extract relevant information. For the users of biological databases, the curated data can be passed to the LLMs to answer user-provided prompts, generating informative outputs. Furthermore, we compared the performance of two pretrained LLMs, GPT-3.5 and GPT-4, showing that improvements in LLMs are likely to contribute to improve performance with the interpretation and biocuration of genetic mapping data.
Materials and methods
Manual curation and display of genetic datasets
To enhance the genetic data content of GrainGenes, with an emphasis on biotic and abiotic stress traits in bread wheat and barley, two collections of journal articles (21 in the first collection and 16 in the second) were manually selected by a curator after a literature review (Supplementary Table S1). These peer-reviewed journal articles contained QTLs and MTAs, such as linkage and genome-wide association study (GWAS) mapping data results for traits, including salinity tolerance, disease resistance, and agronomic traits (Supplementary Table S1). Note that depending on the manuscripts, MTAs can refer to different genetic markers, such as QTLs, quantitative trait nucleotides (QTNs), single-nucleotide polymorphisms (SNPs), simple sequence repeats, and GWAS marker names. Manual curation in GrainGenes involves creating reference records and often creating QTL names to represent the data using existing traits in GrainGenes and previously used abbreviations for the same measured traits. Records include significant MTAs (e.g. locus and probe records), favorable alleles, statistics, and genes when available. Following the community standards, when QTL names are created during curation, the name will contain a “Q,” the trait, a period, a string that describes the population, a dash, and the chromosome followed by a number (e.g. -1D.1, -1D.2, etc.) if there are more than one QTL on the same chromosome for the same trait. When genomic coordinates were provided in a publication that aligned to the IWGSC (International Wheat Genome Sequencing Consortium) Chinese Spring wheat v1.0 assembly [31], these coordinates were used in dedicated genome browser tracks. When coordinates are not provided, the marker sequence is BLASTed (Basic Local Alignment Search Tool) [32] against specific assemblies and the alignment coordinates of the BLAST result are then used to place the QTL on the track. The tracks are available under the “GrainGenes Curated Tracks” menu within the IWGSC Chinese Spring wheat genome browser.
Parsing scientific literature portable document format files and generating vector-store databases
The text of the curated manuscripts was parsed from the portable document format (PDF) files using SciPDF Parser (github.com/titipata/scipdf_parser/), a Python parser for scientific PDFs, and each manuscript was stored as a Python dictionary consisting of section title keys and text content values. The LangChain (www.langchain.com) function RecursiveCharacterTextSplitter was used to split the text from the PDF sections into overlapping chunks, defined by the maximum chunk size (chunk_size) and the overlap size (chunk_overlap) parameters. Specifically, chunk sizes of 250, 500, 750, and 1000 were used, with chunk overlap set to be a tenth of the given chunk size. For each parsed PDF, the OpenAI Application Programming Interface (API) was used to encode all chunks as embedding vectors, stored in individual FAISS (Facebook AI Similarity Search; ai.meta.com/tools/faiss/) vector-store database. To ensure the most relevant information was retrieved from each manuscript for the RAG chatbot, each manuscript was processed and converted to a FAISS database separately. Information presented in tables and figures within the PDFs were not included as part of this study.
Construction of custom retrieval-augmented generation chatbots using LangChain
LangChain is a suite of products that helps developers build and deploy reliable generative AI apps faster. For the tasks involving abstracts, the full abstract texts and OpenAI-encoded embedding vectors were used, with no further modifications. For the tasks of extracting traits, QTLs and MTAs, the RAG approach was used. Specifically, a cosine similarity search of the prompt embedding vector against the manuscript FAISS databases was used to identify the top-k text chunks with the highest cosine similarity scores. The relevant chunks were then retrieved and provided as additional context to the original query, using the LangChain RetrievalQA-based call to the LLM model. For generating structured data from the LLM response, the LLM was tasked with outputting the results in a JavaScript Object Notation (JSON) format that was then parsed by the Python JSON and Pandas packages. For parsing the manually curated structured QTL dataset, the LangChain create_pandas_dataframe_agent function was used to create a chatbot agent. Given a user-specified prompt, the agent was tasked with generating a chain of Python commands to generate the desired outcomes, either as a comma–separated–value-formatted table or a narrated textual response. Two OpenAI models were queried using the OpenAI API, GTP-3.5 (gpt-3.5-turbo-1106), and GPT-4 (gpt-4), using default temperature parameter of 0.7, and their individual performances were assessed and compared.
Performance evaluation
To benchmark the performance of LLMs in the extraction of traits, QTLs, and MTAs directly from the manuscripts, only the subset of traits and MTAs that were correctly extracted were evaluated. To evaluate the performance of GPT in extracting information from structured data and manuscript PDFs, we focused on the percent of correctly extracted information. Because GPT responses for trait and MTA names do not perfectly match the trait and MTA names from the manuscripts, we worked together with a curator to evaluate the accuracy of each response. Incorrectly extracted information was assessed qualitatively, focusing on the general types of mistakes observed. The list of names, responses, and correctness assignments are provided in the supplementary tables.
Results
Developing custom large language model chatbots to assist with curation and information extraction from genetic mapping studies
The diversity of traits studied, mapping populations, and QTL marker naming schemes, mean that access through QTL queries, or the parsing of manually curated structured data requires domain knowledge and computational skills for extracting relevant and useful results. Recent developments in LLMs offer promising opportunities for both extracting trait, QTL, and MTA information from curated data and facilitating the curation process through direct processing of manuscript PDFs [17, 18]. We note that automatic extraction of QTL marker results from tables within PDF files remains challenging and is not considered in the current study. Instead, all the traits, QTLs, and MTAs that were mentioned within the text of the manuscripts were collected to be used as a benchmark. In this study, we consider two main scenarios for using LLMs (i): extracting trait, QTL, and MTA information directly from PDF-formatted manuscripts to assist curators of genetic mapping results and (ii): extracting useful information from manually curated QTL and MTA database to assist users of biological databases based on user-provided prompts (Fig. 1).
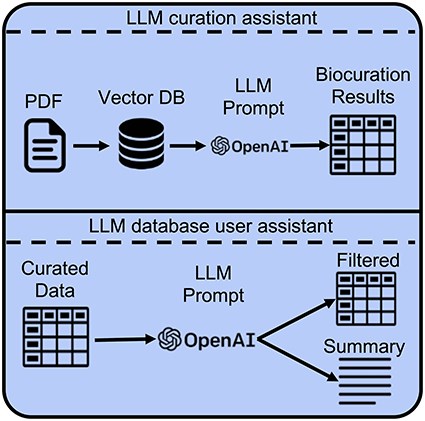
Developing custom LLM-based chatbots to assist with curation and information extraction from genetic mapping studies. Pretrained LLMs, such as the OpenAI GPT models, can be employed to assist both the curators and the users of biological databases. For assistance with curation, LLMs, coupled with the RAG framework, can be used to extract relevant structured data from genetic mapping manuscripts, such as manuscript categories, traits, QTLs, and MTAs. For assistance with manually curated structured QTL data, LLMs can be used to filter and summarize relevant information based on user-provided prompts.
Using large language models for manuscript categorization and grouping
Due to the large body of literature on MTA studies in crops, one obstacle that can be addressed by LLMs is the categorization of manuscripts based on the field of studies and the identification of similar studies for prioritizing manuscripts for curation. During the manual curation of the 36 genetic mapping manuscripts, they were separated into the following broad categorizations: biotic stress, abiotic stress, and agronomic traits (Supplementary Table S1). A prompt for manuscript categorization was generated using the complete abstract as context (Fig. 2a). The OpenAI GPT-3.5 and GPT-4 models were then used to generate a response, prompting the LLM to provide a single, best-fitting, category from the three suggested categories: agronomic, abiotic, and biotic (Fig. 2a). Our results showed that GPT-3.5 and GPT-4 both correctly categorized 97% of the manuscripts (Fig. 2b and Supplementary Table S2). In a second, unsupervised approach, the OpenAI-generated vector embeddings of the 36 abstracts were used to compare manuscripts based on abstract similarity. Using a principal component analysis (PCA) approach, we calculated the first 2 principal components of the 36 abstract vector embeddings. Based on the generated PCA plot, we observed that manuscripts belonging to the three different categories cluster together, and that the 95% confidence interval ellipses do not overlap between the different categories (Fig. 2c).
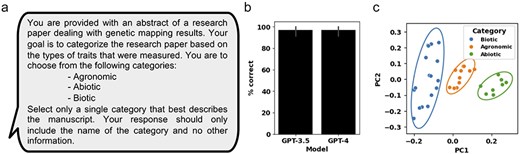
Assessing GPT-3.5 and GPT-4 performance at categorizing genetic mapping manuscripts and using OpenAI LLM-encoding for manuscript similarity comparison. (a) The prompt for OpenAI GPT used to categorize individual manuscripts. (b) The percent of correctly categorized manuscripts using GPT-3.5 and GPT-4 across 36 manuscript abstracts. (c) A PCA plot generated from the OpenAI-encoded vector embeddings of the 36 manuscript abstracts. Manuscripts are colored based on manually curated categories. Ellipses represent the 95% confidence interval for each of the three manually curated categories. Error bars represent standard deviation.
OpenAI generative pre-trained transformer can be used to extract studied traits and conditions from manuscripts
Using the RAG framework, GPT-3.5 and GPT-4 were used to query the FAISS databases of the manuscripts to identify relevant text chunks containing information about the measured traits and conditions used for genetic mapping (Fig. 3a and Supplementary Table S3). Due to variability in results produced, an expert curator manually assessed the accuracy of the extracted traits (Supplementary Tables S4 and S5). The performance accuracy was calculated by comparing the percent of correctly extracted traits from the manually curated traits. Comparing GPT-3.5 and GPT-4 models with top-k of 25 and chunk size of 1000 (see Methods for descriptions of these parameters), we found that GPT-4 performed marginally better than GPT-3.5, correctly extracting an average of 80% and 71% of traits across the 36 manuscripts (Fig. 3b). Next, we assessed two parameters that influence the amount of context provided to the RAG chatbot, top-k size, and chunk size, on Manuscript #2, and had the expert curator assess the generated results (Supplementary Table S5). We found that for both GPT-3.5 and GPT-4, a combination of low chunk size and low top-k size generally has the lowest performance (Fig. 3c and d). For both GPT-3.5 and GPT-4, the best results were achieved with chunk sizes above 750 and top-k sizes higher than 5, with marginally better results for GPT-4 compared to GPT-3.5, correctly extracting an average maximum of 91% and 90% of traits (Fig. 3c and d).
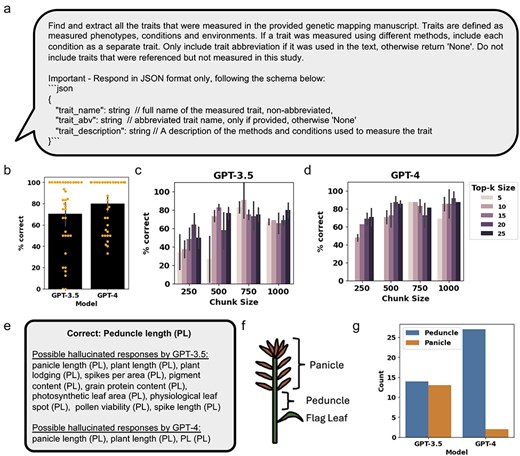
Assessing GPT-3.5 and GPT-4 performance at extracting relevant traits from genetic mapping literature. (a) The prompt for OpenAI GPT used for retrieval of relevant text chunks and generation of a response. (b) Percent of traits correctly extracted for the 36 manuscripts, using top-k size of 25 and chunk size of 1000. Orange dots represent the percent of correctly extracted traits from each of the 21 manuscripts. (c) and (d) Percent of traits correctly extracted from Manuscript #2, using different top-k sizes and chunk sizes for GPT-3.5 and GPT-4, respectively. (e) Examples comparing possible hallucinations observed when assessing the traits extracted by GPT-3.5 and GPT-4. (f) A diagram of the inflorescence characteristic of monocots, such as wheat, showing the relative position of the flag leaf, peduncle, and panicle. (g) Number of times the trait “Peduncle Length” in Manuscript #2 was correctly extracted or possibly altered to “Panicle Length,” across all replicated responses at different top-k sizes and chunk sizes for GPT-3.5 and GPT-4. Error bars represent standard deviation.
In some cases, the LLM produced correct responses but with deviations from the curated trait names. These deviations could be due to a number of possible reasons. In Manuscript #3, GPT-3.5 returned “Race-specific seedling responses” and GPT-4 returned “Stripe rust responses,” while the curated trait name was “stripe rust seedling response,” with all three appearing interchangeably in the manuscript (Supplementary Table S4). In Manuscript #21, the nine curated traits included “ratio” in their name, with both “ratio” and “relative change” being used to describe the same traits, interchangeably. In this case, GPT-3.5 used “relative” in most of the extracted traits names, while GPT-4 used neither (Supplementary Table S4). In other instances, we found that trait names are used interchangeably throughout the text. For example, in Manuscript #16, the curated “well-irrigated conditions” trait was extracted as “low salinity conditions” by GPT-4, both appearing in the manuscript (Supplementary Table S4). Despite being prompted to produce separate trait values, both GPT-3.5 and GPT-4 results can, in some cases, produce aggregate traits. For example, in GPT-3.5, the 10 different mineral traits in Manuscript #10 are combined into a single “Grain mineral concentration” trait, while in GPT-4, the minerals are not aggregated, but only 3 of the 10 minerals have been correctly extracted (Supplementary Table S4).
Among the incorrectly extracted traits, we observed four general types of error (i). The first group consists of predicted traits that were mentioned in the manuscript, often as part of the introduction or discussion. For instance, in Manuscript #7, in addition to the correctly extracted traits, GPT-3.5, but not GPT-4, incorrectly extracts nine traits that were attributed to other genetic mapping manuscripts, and in Manuscript #22, GPT-3.5, but not GPT-4, incorrectly extracted “Seed Vitamin E Content” that was mentioned in the introduction but not measured in the manuscript (Supplementary Table S4) (ii). A second group consists of traits that are not plant phenotypes. For example, in the same manuscript, #7, GPT-4 extracted the traits “Marker-trait associations,” “Broad-sense heritability,” and “Phenotypic variation” (Supplementary Table S4) (iii). A third group consists of traits that were possibly altered by the LLM, possibly through hallucinations. For example, in Manuscript #10, GPT-3.5 possibly altered “Anther retention (AR)” to “Awn length (AR)” and GPT-4 altered “anther extrusion (AE)” to “Anthesis Energy (AE),” with neither of the altered traits appearing in the manuscript (Supplementary Table S4). In the same manuscript, #10, GPT-3.5 possibly altered “Days to anthesis (DA)” to “Disease assessment (DA),” but in this case “disease assessment” appeared elsewhere in the manuscript (Supplementary Table S4) (iv). The fourth type of commonly found mistake is the extraction of traits that can be considered duplications. For example, in the extracted traits from Manuscript #13, for GPT-4, we observed “Leaf blotch disease severity,” “Adult-plant leaf blotch resistance,” and “SNB leaf blotch,” all descriptions from the manuscript that refer to the same leaf blotch disease severity trait (Supplementary Table S4).
In the combined results from GPT-3.5 and GPT-4 models tested with the different top-k size and chunk size parameters on Manuscript #2, we observed repeating cases where, e.g. the peduncle length (PL) trait was incorrectly extracted and hallucinated as a different trait name with the same abbreviation (PL) (Fig. 3e and Supplementary Table S5). More incorrectly extracted traits with the PL abbreviations were observed with GPT-3.5 compared to GPT-4 (Fig. 3e and Supplementary Table S5). While some of the incorrectly extracted traits retained the correct “PL” abbreviation, such as panicle length, plant length, and plant lodging, other incorrectly extracted traits did not, such as spikes per area, pigment content, and pollen viability, despite being extracted with the “PL” abbreviation (Fig. 3e, Supplementary Table S5). Interestingly, PL, the distance between the flag leaf petiole to the base of the panicle (Fig. 3f), was often extracted as panicle length. Comparison of the results for GPT-3.5 and GPT-4 showed that GPT-4 primarily extracted the correct trait name while GPT-3.5 extracted the correct and incorrect trait names at relatively equal amounts (Fig. 3g).
OpenAI generative pre-trained transformer models can be used to extract quantitative trait loci and marker–trait associations from manuscripts
The third LLM task that could be of benefit to the curation process is extracting QTLs and MTAs from manuscripts. Here, MTAs are used as a broad term for genetic markers found to be significantly associated with the studied trait. The engineered prompt for QTL and MTA extraction was written to extract all QTL, QTN, MTA, SNP, and SRR names or GWAS marker names mentioned in the manuscript (Fig. 4a). In addition to the QTL and MTA names, we prompted the GPT models to extract relevant information such as associated traits, chromosomes, and genomic ranges, based on the available information (Fig. 4a). To analyze the LLM performance, we focused on the correctly extracted QTL and MTA names only, with the additional information provided for reference only (Supplementary Table S6). Comparison of the extracted MTAs using the GPT-3.5 and GPT-4 models show that GPT-4 outperformed GPT-3.5, correctly extracting 61% compared to 46% of QTLs and MTAs, respectively (Fig. 4b and Supplementary Table S7). Next, we assessed two parameters that influence the amount of context provided to the RAG chatbot, top-k size, and chunk size, on Manuscript #10, and had the expert curator assess the generated results (Supplementary Table S8). We found that GPT-4 outperformed GPT-3.5 in QTL and MTA extraction in almost all parameter combinations (Fig. 4c and d and Supplementary Table S8). For GPT-3.5, the best results were obtained with chunk size 500 and top-k size of 20, while higher or lower chunk sizes performed poorly with all tested top-k combinations. Furthermore, unlike for GPT-3.5, we observed for GPT-4 that increasing top-k sizes improved QTL and MTA extraction accuracy across all chunk sizes (Fig. 4d and Supplementary Table S8).
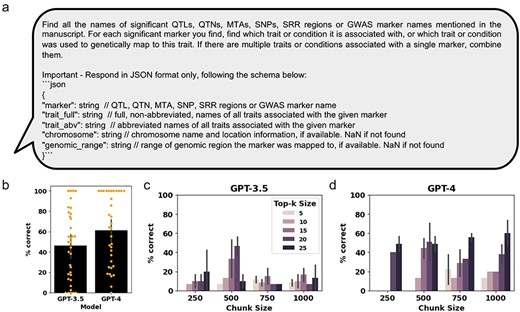
Assessing GPT-3.5 and GPT-4 performance at extracting relevant QTLs and MTAs from genetic mapping literature. (a) The prompt for OpenAI GPT used for retrieval of relevant text chunks and generation of a response. (b) Bar graphs showing the percent of QTLs and MTAs that were correctly extracted for the 36 manually curated manuscripts (Manuscript #14 was excluded from the analysis because no QTLs and MTAs were present in the written portion of the paper). Orange dots represent percent correct traits extracted from each individual manuscript. (c) and (d) Bar graphs showing the percent of traits that were correctly extracted for Manuscript #10, at different chunk sizes and k parameters for GPT-3.5 and GPT-4, respectively. Error bars represent standard deviation.
Similar to the extracted traits, the LLMs produced in some cases correct responses but with deviations from the curated QTL and MTA names. For example, the QTLs and MTAs extracted by GPT-3.5 for Manuscript #6 were superseded with “SNP,” despite it not being part of their name (Supplementary Table S7). Additionally, Manuscripts #10 and #17, GPT-4 but not GPT-3.5, grouped a number of QTLs and MTAs as comma separated values (Supplementary Table S7). Among the incorrectly extracted traits, we observed four general types of error (i): In Manuscript #18, instead of QTLs and MTAs, the gene symbols Rht-B1, Rht-D1, and Rht8 were mistakenly extracted by GPT-3.5 (but not by GPT-4) (Supplementary Table S7) (ii). In Manuscript #2, GPT-3.5, but not GPT-4, mistakenly extracted trait names instead of QTL and MTA names (Supplementary Table S7) (iii). In Manuscript #14, when the manuscript text did not include any QTLs or MTAs (Supplementary Table S6), both GPT-3.5 and GPT-4 provided wrong output about some genomic regions that were mentioned in the manuscript (Supplementary Table S7). Compared to the trait extraction results, both GPT-3.5 and GPT-4 had little hallucinations for the QTLs and MTAs that were correctly extracted (Supplementary Table S7).
GPT-4, but not GPT-3.5, can be used to efficiently retrieve information from manually curated data
The manually curated QTLs hosted in the GrainGenes database contain additional information, such as genomic coordinates, associated traits, and the MTAs used to define each QTL. While the task of finding QTLs in a given genomic region is relatively straightforward, the task of finding QTLs related to specific types of traits can be more challenging due to the variety of traits studied (Supplementary Table S9). Although not specifically designed for this, pretrained LLMs have been adapted to effectively interact with tabular data [33]. To assess the ability of LLMs in extracting relevant QTLs, we aggregated all the QTLs from the 21 manually curated genetic mapping manuscripts (Supplementary Table S9). Specifically, we tested the ability of LLMs to retrieve QTLs associated with response to disease or disease resistance traits, either across the full genome or within a particular genomic range (Fig. 5a). We observed that GPT-3.5 failed to correctly extract relevant data with the minimal prompt for both the full and ranged data, but, on average, correctly extracted 62% and 33% of the QTLs given the additional instructions for the full and ranged data, respectively (Fig. 5b and Supplementary Table S10). GPT-4, on average, correctly extracted 29% and 31% of the QTLs with the minimal prompt for the full and ranged data, respectively, and 91% and 96% with the additional instructions, for the full and ranged data, respectively (Fig. 5c and Supplementary Table S10).
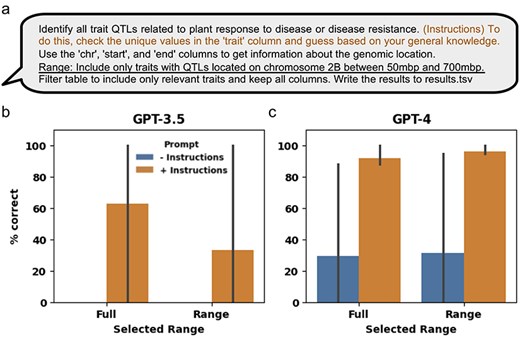
Using GPT-3.5 and GPT-4 to extract information from manually curated QTL and MTA structured data. (a) The minimal prompt and added engineered prompt (in orange) providing additional instructions used for extracting relevant QTL and MTA data from the aggregation of manually curated genetic mapping data of 21 manuscripts. The data were extracted either from the full dataset or using the added “Range” prompt (underlined) to generate the ranged response. (b) and (c) Percent of correctly extracted QTLs by the LLM using the minimal (“− instructions”) and engineered (“+ instructions”) prompts generated by the OpenAI GPT-3.5 and GPT-4 LLM models, respectively. Error bars represent standard deviation.
GPT-4, but not GPT-3.5, can be used to generate a narrated textual summary from manually curated data
In addition to extracting relevant information from tabular data, LLMs can be used to generate an output that provides a narrated textual summary of the requested information. Specifically, we tested the ability of LLMs to generate a narrated textual summary from the retrieved QTLs associated with response to disease or disease resistance traits, either across the full genome or within a particular genomic range (Fig. 6a). We observed that response lengths for GPT-3.5, on average, were 289 and 149 characters long with the minimal prompt for the full and ranged data, respectively, and 174 and 169 characters long with the additional instructions, for the full and ranged data, respectively (Fig. 6b). The response lengths for GPT-4, on average, were 546 and 629 characters long with the minimal prompt for the full and ranged data, respectively, and 1290 and 692 characters long with the additional instructions, for the full and ranged data, respectively (Fig. 6c). A representative response generated for the ranged response query for both LLM models showed that GPT-3.5 failed to extract relevant QTLs, while GPT-4 was able to generate a narrated textual summary (Fig. 6d).
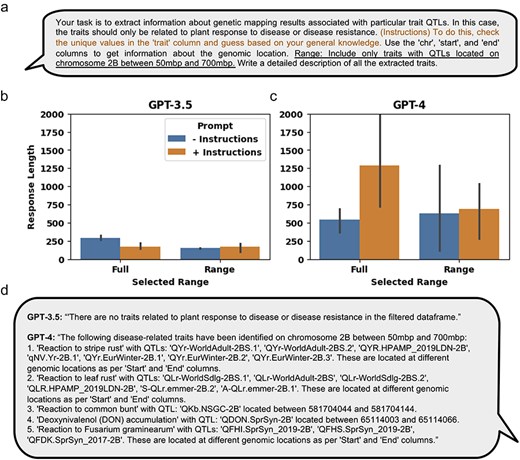
Using GPT-3.5 and GPT-4 to extract information and write a narrative from manually curated QTL and MTA structured data. (a) The minimal prompt and added engineered prompt (in orange) providing additional instructions used for extracting relevant QTL and MTA data from the aggregation of manually curated genetic mapping data of 21 manuscripts. The data were extracted either from the full dataset or using the added “Range” prompt (underlined) to generate the ranged response. (b) and (c) Results of the response lengths generated by GPT-3.5 and GPT-4, respectively, either on the full dataset or a selected range. Response lengths do not necessarily mean accurate results, but it is a measure of output that may affect the amount of information content. (d) An example response generated by either GPT-3.5 or GPT-4 using the engineered prompt and within the selected range. Error bars represent standard deviation.
Discussion
Curation of genetic mapping literature involves the manual extraction of QTL and MTA data from manuscripts and integrating the data with easily accessible genomic databases [30]. The primary goal for curating MTAs is to provide a centralized location for genomic loci associated with different traits, facilitating both breeding and research efforts [30]. While LLMs have shown great potential in automating information extraction tasks for different biological data types, such as in medicine, gene ontology, and chemical reactions [9, 21–24], their ability to automate analyses of genetic mapping literature remains untested. Here, we assess the utility and limitations of using LLMs for assisting curators and users of biological databases in tasks related to genetic mapping literature. From the standpoint of curators, we assessed the following three tasks: (i) manuscript categorization, (ii) trait extraction, and (iii) QTL and MTA extraction. From the users’ standpoint, we assessed the following two tasks: (i) extraction of relevant data based on user-specified instructions and (ii) generation of summarized narrated responses from manually curated QTLs and MTAs. The lack of standardized nomenclature contributes to the need for professionally trained manual curators and for a community effort to generate standardized nomenclature and reporting procedures [1, 2]. A combined effort by the research community, funding agencies, and open-access journals to add open data requirements is likely to simplify the curation process and increase the availability of research outcomes [2]. This will also likely have the beneficial effect of improving performance when analyzing literature and research outcomes using LLMs.
Grouping genetic mapping literature based on the similarity of studied traits facilitates comparative analysis of significantly associated genomic loci and markers to help prioritize breeding and research efforts [2]. The first task we considered was using LLMs to categorize genetic mapping manuscripts based on broad categories. In our study of 36 genetic mapping studies, the manually curated QTLs were separated into 3 overarching categories: agronomic, abiotic, and biotic traits. Because abstracts provide a succinct summary of the study, are consistently formatted, and easily accessible, we focused on applying LLMs solely on abstracts for manuscript categorization. When tasked with selecting the most appropriate category of the three provided categories, both GPT-3.5 and GPT-4 performed well and consistently, with only Manuscript #16 being incorrectly categorized as an agronomic-related manuscript, despite being curated as an abiotic category manuscript (Supplementary Table S2). The abstract of Manuscript #14 described the study of agronomic traits under different salinity levels, suggesting that manuscript categorization should allow for multi-category predictions. In the second approach, we compared abstract similarity using the OpenAI-generated vector embeddings on the full abstract texts. Our findings demonstrate that the first two principal components (PC1 and PC2) effectively separate the 36 manuscripts into distinct groups that align with the manually curated categories (Fig. 2c). Thus, GPT-3.5 and GPT-4 can be used to effectively predict general categories and cluster manuscripts based on vector embedding similarity using abstracts alone, with both approaches being easily scalable to large numbers of manuscripts (Fig. 2b and c).
Next, we considered using LLMs to facilitate the process of manual curation of genetic mapping literature by directly extracting information from manuscripts. At the task of extracting trait information, we observed that GPT-4 outperforms GPT-3.5, correctly extracting 80% and 71% of traits, respectively, across the 36 manuscripts (Fig. 3b). When comparing the effect of the top-k size and chunk size parameters on extracting traits from Manuscript #2, GPT-4 generally outperformed GPT-3.5 and benefited more than GPT-3.5 when more text chunks were provided (Fig. 3c and d). Unlike trait names, QTLs and MTAs designations are often unique to individual manuscripts and tend not to follow strict nomenclature, suggesting that longer contextual information can improve QTL and MTA extraction by LLMs [2]. The markers found within the text are often limited to a select few that the authors considered to be of higher relevance and importance, and even this observation can be highly context-dependent. Here, GPT-4 outperformed GPT-3.5, correctly extracting 61% compared to 46% of QTLs and MTAs, respectively (Fig. 4b). Additionally, GPT-4 outperformed GPT-3.5 in almost all top-k size and chunk size parameter combinations, with an increase to the top-k size parameter providing the most substantial performance improvement across the different chunk sizes (Fig. 4c and d). The apparent advantage of GPT-4 over GPT-3.5 to more accurately extract traits, QTLs, and MTAs when given more information could be explained by the larger model size (trained on 170 trillion tokens for GPT-4 and 175 billion tokens for GPT-3.5) and larger context window length (8192–32 768 for GPT-4 and 2048 for GPT-3.5) [34].
In addition to extracting data from manuscripts, LLMs can also be used to extract relevant information from structured data. In this case, we used a dataset of manually curated QTLs from the first collection of 21 manuscripts that were recently deposited in the GrainGenes database. The QTLs span across the wheat genome and encompass different traits, including agronomic, abiotic and biotic traits (Supplementary Tables S1 and S9). Curated QTL results are a valuable resource for biologists, geneticists, and breeders and extracting relevant information often requires domain knowledge that can be a time-consuming effort. Here, we consider using pretrained LLMs as a tool to easily extract useful information from a large set of curated QTLs based on user-provided instructions. Our results suggest that GPT-3.5 produces low-quality and inconsistent results when tasked with extracting relevant QTLs from manually curated QTL-structured data (Fig. 5b and c). On the other hand, GPT-4 is able to produce high-quality and consistent results, although that specific outcome required prompt engineering that provided additional instructions to the LLM on how to process the data (Fig. 5a and c). Thus, when the engineered prompt provided the instruction: “To do this, check the unique values in the “trait” column and guess based on your general knowledge.” GPT-4 was able to assess all individual trait names for their relation to response to disease or disease resistance, instead of just searching for the generic “disease” or “resistance” keywords in the trait names. For example, by considering each trait separately, GPT-4 correctly extracted QTLs associated with a wheat fungal pathogen (reaction to common bunt, Fusarium graminearum, and stripe rust) and mycotoxin accumulation (deoxynivalenol accumulation) traits, correctly inferring their association with response to disease or disease resistance while reducing the need for domain knowledge (Supplementary Table S9). Similar outcomes were observed when tasking LLMs with generating narrated responses from manually curated data, with GPT-4 producing more accurate and detailed responses than GPT-3.5, even though both failed to provide a detailed explanation of the observed traits (Fig. 6b and d).
One major challenge is the assessment of the accuracy of LLMs in extracting and processing genetic mapping data. In this work, all results generated by the LLMs were manually assessed by an expert curator, requiring a significant time investment. Because assessing the accuracy of the results generated by LLMs requires an extensive manual effort, only two LLM models (GPT-3.5 and GPT-4), two parameters (top-k and chunk size), and one case of prompt engineering were assessed in this study. To facilitate further improvements in applying LLMs to genetic mapping results, it is worth considering a community effort to compile a comprehensive benchmark dataset across different plant species with a variety of genetic mapping manuscripts [35]. This would enable a more comprehensive testing of different LLM models, parameters, and prompt engineering approaches, leading to improved performance [36]. Furthermore, it is likely that a one-fits-all approach will be hard to develop. For example, trait numbers range from 1 in Manuscript #9 to 17 in Manuscript #20, likely requiring substantially different context for the LLMs to extract all relevant data. Recently developed pretrained LLM models with long-context capabilities might negate the need for using RAG and tuning parameters such as top-k size and chunk size, by passing whole documents to the LLMs [16]. However, research suggests that pipelines combining RAG and long-context LLMs can better leverage advancements in LLMs and provide improved performance while reducing cost and computational resources [37–39]. Improvements in RAG systems and workflows that enhance retrieval accuracy and robustness in complex cases could further improve LLM-enabled biocuration methods [40, 41]. Our investigation highlighted the necessity of manual curation for each manuscript due to inconsistent formatting, emphasizing the relevance of standardizing reports of genetic mapping results data. Comparative analysis revealed that GPT-4 outperformed GPT-3.5 across various tasks, indicating that continual improvement of LLMs will lead to further improvements in tasks relating to extraction of genetic mapping data from manuscripts and manually curated data.
Conclusion
Lower experimental and computational costs are driving the accelerated increase in the amount of generated biological data, creating bottlenecks in data intake and curation, especially for community biological databases. The limiting factor for bringing in more data and linking each data point to other data types is the number of domain experts dedicated to curation. The disruptive increase in performance recently demonstrated by generative AI technologies, particularly with LLMs, points to their immense potential to facilitate biocuration and help users of community databases access and use data in their research. In this manuscript, we used a small set of scientific journal articles to evaluate the ability of GPT to extract relevant information from genetic mapping literature and manually curated data through precise prompting. Although our dataset is limited to 36 journal articles, our results demonstrated that GPT models are highly capable of extracting and presenting genetic and genomic information accurately, when mindful prompting is exercised. Our results show that GPT-4 provides consistently better results than GPT-3.5, suggesting that further improvements in LLMs will have a significant impact on assisting human curators and as an interface for everyday biocuration database users. We also show that a certain level of caution should be taken due to incorrect responses and the implications of LLM hallucinations.
Acknowledgements
Mention of trade names or commercial products is solely for the purpose of providing specific information and does not imply recommendation or endorsement by the USDA. USDA is an equal opportunity provider and employer. All URLs were accessed in March 2024.
Supplementary data
Supplementary data are available at Database Online.
Conflict of interest:
None declared.
Funding
This research was supported by the U.S. Department of Agriculture, Agricultural Research Service, Project Numbers 2030-21000-056-00D and 5030-21000-072-000D through the Crop Improvement and Genetics Research and Corn Insects and Crop Genetics Research Units.
Data Availability Statement
The data underlying this article are available in its online supplementary material and in GitHub, at https://github.com/eporetsky/ChatMTA.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}