Abstract
Human POLE and POLD1 genes encode DNA polymerases responsible for genome replication and proofreading of DNA synthesis errors. Germline and somatic POLE/POLD1 mutations compromising the polymerase fidelity cause cancers with high mutational burden. Ultramutation is associated with a better prognosis and immunotherapy response, highlighting the need to define tumour POLE/POLD1 status unambiguously. Prior studies assessed the functional significance of numerous POLE/POLD1 variants in experimental models. However, the data remain scattered and difficult to evaluate by non-specialists, limiting their utility for research and clinical applications. Through manual literature curation, we integrated data from functional studies of clinically relevant POLE and POLD1 variants into PolED, a publicly available database (https://poled-db.org). PolED compiles information on variant effects in biochemical assays, yeast, mammalian cells, and mouse tumour models along with supporting references. It also includes a concise summary of functional significance for each variant. PolED aims to assist in clinical decision-making, guide personalized therapy, and promote further research.
Introduction
Genome stability relies on multiple mechanisms, including DNA repair pathways, dNTP pools maintenance, accurate DNA replication, and cell cycle checkpoints. Among these, high-fidelity DNA synthesis by replicative polymerases ε (Pol ε) and δ (Pol δ) is the most critical for preventing spontaneous mutations [1, 2]. The catalytic subunits of Pol ε and Pol δ, encoded by the POLE and POLD1 genes in humans, contain a DNA polymerase domain for accurate processive DNA synthesis and an exonuclease domain for correcting rare misincorporation errors. Mutations affecting nucleotide selection by the polymerase domain or proofreading by the exonuclease domain reduce the accuracy of DNA synthesis, increase mutagenesis, and cause cancer predisposition in experimental models [3–17].
Over 12 000 non-silent POLE/POLD1 variants have been reported in humans (Supplementary Tables S1–S3). A subset of these variants impair the polymerase fidelity and drive the development of tumours with exceptionally high mutation burdens. Somatic POLE/POLD1 mutations occur across many cancer types [18], but are particularly frequent in colorectal and endometrial cancer [19, 20] and in brain tumours of children with constitutional DNA mismatch repair deficiency (CMMRD) [21, 22]. Germline POLE/POLD1 mutations cause high-penetrance adult-onset cancer predisposition syndromes [23–26] and have also been associated with paediatric malignancies [27–33]. Ultramutated tumours exhibit improved outcomes due to high neoantigen production and an enhanced anti-tumour immune response. They also respond well to immune checkpoint inhibitors [34, 35]. Thus, POLE/POLD1 mutation status is a promising predictive biomarker for immunotherapy treatment. Additionally, detecting germline POLE/POLD1 drivers is crucial for risk assessment and long-term patient management in families with the hereditary syndromes [36, 37].
Distinguishing driver POLE/POLD1 alleles and benign variants is not straightforward. Pol ε and Pol δ are essential for DNA replication. Mutations result from error-prone DNA synthesis by these enzymes; therefore, only missense variants that preserve robust DNA synthesis capacity could theoretically drive ultramutation. Loss-of-function variants (deletions, insertions, premature stop codons) prevent production of the enzyme and cannot cause ultramutated cancers. The functional impact of most missense variants—except those at catalytic amino acid residues—is difficult to predict without experimental analysis. Since the discovery of Pol ε and Pol δ genes in the late 1980s [38–40], structure-function studies and genetic screens identified many amino acid substitutions that disrupt polymerase fidelity and increase mutation rates [4–7, 10, 12, 13, 15, 16, 41–57]. Some of this information was instrumental in establishing the pathogenicity of first reported human variants. The discovery of a multitude of POLE/POLD1 mutations in human cancers in the 2010s further boosted functional analysis efforts, which now specifically focused on cancer-associated variants with suspected clinical significance [22, 23, 58–85]. Due to the high conservation of polymerase functional regions, yeast model systems have been used extensively to engineer mutations analogous to human variants and determine their impact on the mutation rate. In vitro assays with purified Pol ε and Pol δ helped understand the effects of the variants on exonucleolytic proofreading. Some variants have been engineered in cultured human cells and mice to assess the impact on mutation rates, tumour susceptibility, and immunotherapy response. The availability of functional analysis data remains an important consideration in gauging the pathogenicity of new variants. However, much of the available experimental data on POLE/POLD1 variants remains unconsolidated. In the early literature, studies of the polymerase variants do not typically discuss the link to cancer, making these data even more difficult to find. Moreover, interpreting experimental data on mutator phenotypes and DNA polymerase fidelity requires specialized expertise not easily available to clinicians or interdisciplinary researchers.
To address these issues, we developed PolED, a manually curated database that compiles functional studies on POLE and POLD1 variants reported in humans. PolED consolidates and structures data from diverse experimental systems—purified proteins, yeast, cultured mammalian cells, and mouse models—to aid in variant classification. The data are reviewed for reliability and presented in a user-friendly format, making them accessible to non-specialists. PolED assists clinicians in making patient management decisions. It also helps researchers navigate through previously investigated variants and design further studies (Fig. 1).
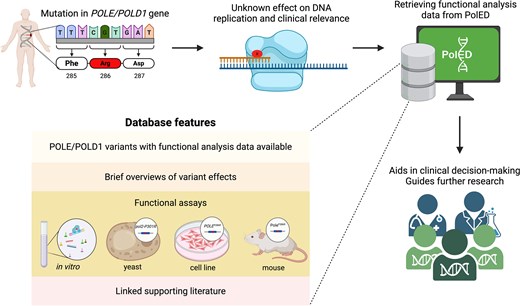
Figure 1.
PolED features and application in variant interpretation. Most POLE/POLD1 mutations are classified as variants of unknown significance. PolED compiles functional analysis data on POLE/POLD1 variants reported in humans. This information can assist clinicians and researchers in interpreting clinical variants.
Data collection and curation
We assessed 12 820 variants (8736 POLE and 4084 POLD1; Supplementary Tables S1 and S2) for the availability of functional analysis data. We extracted these variants from cBioPortal, gnomAD, ClinVar, COSMIC, Human Gene Mutation Database (HGMD), The Single Nucleotide Polymorphism Database (dbSNP), Leiden Open Variation Database (LOVD) v.3.0, OncoKB, Cancer Cell Line Encyclopedia (CCLE), and an internal Shcherbakova laboratory literature database maintained since 1990 and cross-checked using PubMed searches using the terms ‘POLE’ and ‘mutation’, or ‘POLD1’ and ‘mutation’ (Fig. 2A; Supplementary Table S3). Most were missense variants, and ∼1.5% were in-frame deletions or insertions. Among the 8736 POLE variants, 2204 were reported both in the germline and as somatic mutations in tumours, 5872 only as germline variants, and 660 only as somatic variants. Among the 4084 POLD1 variants, 1180 were reported in both germline and somatic contexts, 2480 only as germline variants, and 424 only as somatic variants (Fig. 2B).
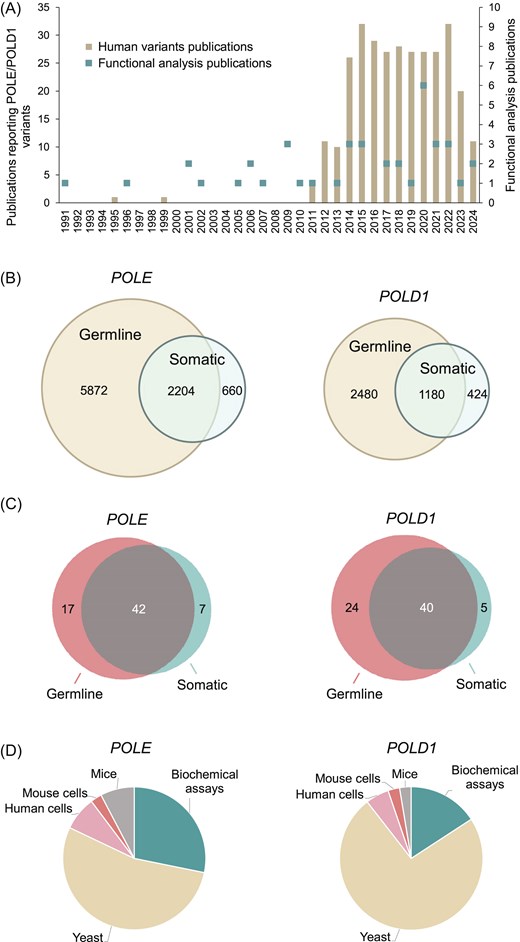
Figure 2.
Literature curation and database content. (A) Publications reporting human POLE/POLD1 variants and their functional studies in experimental models. See Supplementary Tables S3 and S4 for the complete list of publications. (B) Venn diagrams of germline and somatic POLE/POLD1 variants reported in the literature and public databases as of August 2024. See Supplementary Tables S1 and S2 for the complete list of variants. (C) Venn diagrams of POLE/POLD1 variants, for which functional analysis data are included in PolED. (D) Summary of PolED data by experimental model.
To collect functional analysis data, we manually curated 90 publications reporting studies of Pol ε and Pol δ variants in experimental assays. We extracted these publications, dating from 1991 to 2025, from the Shcherbakova laboratory literature database cross-checked via extensive PubMed searches. From this literature, we selected data on the variants reported in humans. The data underwent a thorough review for evidence of scientific rigour and statistical significance before inclusion in PolED. At this time, PolED references only original publications reporting a variant’s effect for the first time. Subsequent studies repeating the analysis are cited only if they add new information (e.g. the phenotype of heterozygous vs. homozygous cells, biochemical effects on different polymerase subcomplexes, or tissue-specific vs. whole-body knock-ins in mice). Figure 2A and Supplementary Table S4 show functional analysis publications included in the current version of PolED. In rare cases, PolED includes unpublished data from our laboratory if there are no analogous published reports. In these cases, the experimental assays use the same methodology and adhere to the same quality standards as our published work. These data are available upon request.
The current PolED version contains functional analysis data for 67 POLE and 69 POLD1 variants. Most occurred both in the germline and as somatic mutations in tumours. Seventeen POLE and 24 POLD1 variants were reported only in the germline, and seven POLE and five POLD1 variants were reported only as somatic mutations (Fig. 2C). Experimental models include in vitro biochemical assays with purified polymerases, in vivo mutation rate assays in yeast Saccharomyces cerevisiae or Schizosaccharomyces pombe, ex vivo mutation rate assays in cultured human or mouse cells, and in vivo mouse models (Fig. 2D). PolED also provides functional information on 32 catalytic amino acid residue variants with no direct experimental data, because structural and functional studies of other polymerases established the importance of these residues for catalysis of the exonuclease reaction.
Database design and features
The PolED web application is developed using Uvicorn (0.34.1, an ASGI server) and FastAPI (v0.115.12, a Python web framework). It runs in a Python 3.10.12 virtual environment and is managed by systemd on a Linux server (Fig. 3, top). Variants and associated information are stored in SQLite (3.37.2) and accessed via SQLAlchemy (2.03.23). The interface is rendered using HTML, CSS, and JavaScript with static content managed by FastAPI. Visual design is powered by Bootstrap (v5.3.5). The interface is adapted for both desktop and mobile browsers. Processing scripts are developed in Python and JavaScript. PolED supports access via modern web browsers, including Chrome, Firefox, and Safari.
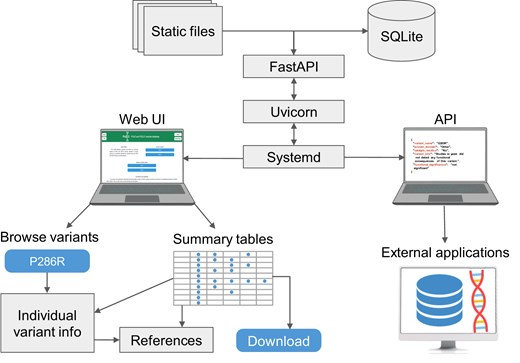
Figure 3.
PolED architecture and interface. The PolED database is implemented as a FastAPI-based web application, deployed using Uvicorn, and managed via systemd. The backend stores variant data in an SQLite database and serves both static files and dynamic content. Users can interact with PolED through a web-based interface or programmatically via the API. The interface allows users to explore data on individual variants or view and download summary tables of all variants with demonstrated functional significance. The API enables integration with external tools and workflows (e.g. NGS pipelines, annotation tools, external databases).
Users access the PolED database through an intuitive web interface. It includes options to explore data on individual POLE/POLD1 variants through the variant browser or view and download summary tables of all variants with demonstrated functional significance (Fig. 3, bottom left). Variants with evidence of functional significance are marked by colour in the variant browser (blue = significant; grey = not significant). Each variant has a dedicated page with a brief summary of functional effects and links to detailed summaries of available data from specific experimental systems (biochemical assays, yeast, human cells, mouse cells, or mouse tumour models). Both the variant-specific pages and the summary tables provide hyperlinks to the corresponding literature references. Users can contribute new functional analysis data through the “Submit data” page.
PolED also provides a representational state transfer (REST) application programming interface (API) (Fig. 3, bottom right). The API is documented and can be examined in an interactive Swagger UI interface. The API retrieves data in JavaScript Object Notation (JSON) format and supports the following two endpoints:
Conclusions and future developments
In this report, we introduce PolED, a web-based database that allows clinicians and researchers to access and analyse functional data on POLE and POLD1 variants in a user-friendly format. By consolidating experimental findings from diverse model systems and presenting them in a structured way, PolED addresses the current gap in the functional annotation of these clinically relevant variants. It emphasizes variant effects on properties relevant to the development of ultramutated cancers: exonuclease and polymerase activities, DNA synthesis fidelity, mutation rate, and tumour susceptibility. The resource facilitates variant interpretation and aids in clinical decision-making, particularly in cancer patient management, where ultramutated phenotypes have emerged as biomarkers for immunotherapy. PolED also helps interpret germline POLE and POLD1 variants to facilitate risk assessment and surveillance of families with cancer predisposition syndromes. Additionally, PolED stimulates further studies of ultramutation in cancer by systematically organizing existing information and providing a catalogue of alleles available as research tools.
We expect that the number of reported POLE and POLD1 variants will grow, and more variants will be examined in experimental assays. We will continue to evaluate new findings and incorporate them into PolED. Data discovered through literature searches or submitted by users are manually curated, processed, and incorporated into the database release within a week of discovery or submission. These data will be of interest to both clinicians and a broad community of researchers in the fields of genome instability and cancer biology.
Acknowledgements
We thank Maggie Luong, Stephanie Barbari, Dmitry Gordenin, Doug Levine, and Diego Castrillon for valuable feedback on the database.
Conflict of interest
None declared.
Funding
This work was supported by the National Institutes of Health grants CA239688 and ES015869 to P.V.S.
Data availability
PolED primarily compiles published data and is freely available at https://poled-db.org/. Unpublished data included in the database are available upon reasonable request to the corresponding author.
References
1.Kunkel
TA
.
DNA replication fidelity
.
J Biol Chem
.
2004
;
279
:
16895
–
98
.
2.Kunkel
TA
.
Evolving views of DNA replication (in)fidelity
.
Cold Spring Harb Symp Quant Biol
.
2009
;
74
:
91
–
101
.
3.Albertson
TM
, Ogawa
M
, Bugni
JM
et al.
DNA polymerase ε and δ proofreading suppress discrete mutator and cancer phenotypes in mice
.
Proc Natl Acad Sci USA
.
2009
;
106
:
17101
–
4
.
4.Fazlieva
R
, Spittle
CS
, Morrissey
D
et al.
Proofreading exonuclease activity of human DNA polymerase δ and its effects on lesion-bypass DNA synthesis
.
Nucleic Acids Res
.
2009
;
37
:
2854
–
66
.
5.Fortune
JM
, Stith
CM
, Kissling
GE
et al.
RPA and PCNA suppress formation of large deletion errors by yeast DNA polymerase δ
.
Nucleic Acids Res
.
2006
;
34
:
4335
–
41
.
6.Ghodgaonkar
MM
, Kehl
P
, Ventura
I
et al.
Phenotypic characterization of missense polymerase-δ mutations using an inducible protein-replacement system
.
Nat Commun
.
2014
;
5
:
4990
.
7.Goldsby
RE
, Hays
LE
, Chen
X
et al.
High incidence of epithelial cancers in mice deficient for DNA polymerase δ proofreading
.
Proc Natl Acad Sci USA
.
2002
;
99
:
15560
–
5
.
8.Goldsby
RE
, Lawrence
NA
, Hays
LE
et al.
Defective DNA polymerase-δ proofreading causes cancer susceptibility in mice
.
Nat Med
.
2001
;
7
:
638
–
9
.
9.Korona
DA
, Lecompte
KG
, Pursell
ZF
.
The high fidelity and unique error signature of human DNA polymerase ε
.
Nucleic Acids Res
.
2011
;
39
:
1763
–
73
.
10.Li
L
, Murphy
KM
, Kanevets
U
et al.
Sensitivity to phosphonoacetic acid: a new phenotype to probe DNA polymerase δ in Saccharomyces cerevisiae
.
Genetics
.
2005
;
170
:
569
–
80
.
11.Morrison
A
, Bell
JB
, Kunkel
TA
et al.
Eukaryotic DNA polymerase amino acid sequence required for 3'-→5' exonuclease activity
.
Proc Natl Acad Sci USA
.
1991
;
88
:
9473
–
7
.
12.Pursell
ZF
, Isoz
I
, Lundstrom
EB
et al.
Yeast DNA polymerase ε participates in leading-strand DNA replication
.
Science
.
2007
;
317
:
127
–
30
.
13.Schmitt
MW
, Matsumoto
Y
, Loeb
LA
.
High fidelity and lesion bypass capability of human DNA polymerase δ
.
Biochimie
.
2009
;
91
:
1163
–
72
.
14.Shcherbakova
PV
, Pavlov
YI
, Chilkova
O
et al.
Unique error signature of the four-subunit yeast DNA polymerase ε
.
J Biol Chem
.
2003
;
278
:
43770
–
80
.
15.Simon
M
, Giot
L
, Faye
G
.
The 3' to 5' exonuclease activity located in the DNA polymerase δ subunit of Saccharomyces cerevisiae is required for accurate replication
.
EMBO J
.
1991
;
10
:
2165
–
70
.
16.Venkatesan
RN
, Hsu
JJ
, Lawrence
NA
et al.
Mutator phenotypes caused by substitution at a conserved motif A residue in eukaryotic DNA polymerase δ
.
J Biol Chem
.
2006
;
281
:
4486
–
94
.
17.Venkatesan
RN
, Treuting
PM
, Fuller
ED
et al.
Mutation at the polymerase active site of mouse DNA polymerase δ increases genomic instability and accelerates tumorigenesis
.
Mol Cell Biol
.
2007
;
27
:
7669
–
82
.
18.Cerami
E
, Gao
J
, Dogrusoz
U
et al.
The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data
.
Cancer Discov
.
2012
;
2
:
401
–
4
.
19.Cancer Genome Atlas Network.
Comprehensive molecular characterization of human colon and rectal cancer
.
Nature
.
2012
;
487
:
330
–
7
.
20.Cancer Genome Atlas Research Network
.
Integrated genomic characterization of endometrial carcinoma
.
Nature
.
2013
;
497
:
67
–
73
.
21.Erson-Omay
EZ
, Cağlayan
AO
, Schultz
N
et al.
Somatic POLE mutations cause an ultramutated giant cell high-grade glioma subtype with better prognosis
.
Neuro Oncol
.
2015
;
17
:
1356
–
64
.
22.Shlien
A
, Campbell
BB
, de Borja
R
et al.
Combined hereditary and somatic mutations of replication error repair genes result in rapid onset of ultra-hypermutated cancers
.
Nat Genet
.
2015
;
47
:
257
–
62
.
23.Palles
C
, Cazier
JB
, Howarth
KM
et al.
Germline mutations affecting the proofreading domains of POLE and POLD1 predispose to colorectal adenomas and carcinomas
.
Nat Genet
.
2013
;
45
:
136
–
44
.
24.Rohlin
A
, Zagoras
T
, Nilsson
S
et al.
A mutation in POLE predisposing to a multi-tumour phenotype
.
Int J Oncol
.
2014
;
45
:
77
–
81
.
25.Valle
L
, Hernandez-Illan
E
, Bellido
F
et al.
New insights into POLE and POLD1 germline mutations in familial colorectal cancer and polyposis
.
Hum Mol Genet
.
2014
;
23
:
3506
–
12
.
26.Vande Perre
P
, Siegfried
A
, Corsini
C
et al.
Germline mutation p.N363K in POLE is associated with an increased risk of colorectal cancer and giant cell glioblastoma
.
Fam Cancer
.
2019
;
18
:
173
–
78
.
27.Berrino
E
, Filippi
R
, Visintin
C
et al.
Collision of germline POLE and PMS2 variants in a young patient treated with immune checkpoint inhibitors
.
NPJ Precis Oncol
.
2022
;
6
:
15
.
28.Lindsay
H
, Scollon
S
, Reuther
J
et al.
Germline POLE mutation in a child with hypermutated medulloblastoma and features of constitutional mismatch repair deficiency
.
Cold Spring Harb Mol Case Stud
.
2019
;
5
:
a004499
.
29.Michaeli
O
, Ladany
H
, Erez
A
et al.
Di-genic inheritance of germline POLE and PMS2 pathogenic variants causes a unique condition associated with pediatric cancer predisposition
.
Clin Genet
.
2022
;
101
:
442
–
47
.
30.Schamschula
E
, Kinzel
M
, Wernstedt
A
et al.
Teenage-οnset colorectal cancers in a digenic cancer predisposition syndrome provide clues for the interaction between mismatch repair and polymerase δ proofreading deficiency in tumorigenesis
.
Biomolecules
.
2022
;
12
.
31.Sehested
A
, Meade
J
, Scheie
D
et al.
Constitutional POLE variants causing a phenotype reminiscent of constitutional mismatch repair deficiency
.
Hum Mutat
.
2022
;
43
:
85
–
96
.
32.Tesi
B
, Lagerstedt-Robinson
K
, Abel
F
et al.
Diagnostic yield and clinical impact of germline sequencing in children with CNS and extracranial solid tumors—a nationwide, prospective Swedish study
.
Lancet Reg Health Eur
.
2024
;
39
:
100881
.
33.Wimmer
K
, Beilken
A
, Nustede
R
et al.
A novel germline POLE mutation causes an early onset cancer prone syndrome mimicking constitutional mismatch repair deficiency
.
Fam Cancer
.
2017
;
16
:
67
–
71
.
34.Kogl
J
, Pan
TL
, Marth
C
et al.
The game-changing impact of POLE mutations in oncology-a review from a gynecologic oncology perspective
.
Front Oncol
.
2024
;
14
:
1369189
.
35.Ma
X
, Dong
L
, Liu
X
et al.
POLE/POLD1 mutation and tumor immunotherapy
.
J Exp Clin Cancer Res
.
2022
;
41
:
216
.
36.Mur
P
, Viana-Errasti
J
, Garcia-Mulero
S
et al.
Recommendations for the classification of germline variants in the exonuclease domain of POLE and POLD1
.
Genome Med
.
2023
;
15
:
85
.
37.Palles
C
, Martin
L
, Domingo
E
et al.
The clinical features of polymerase proof-reading associated polyposis (PPAP) and recommendations for patient management
.
Fam Cancer
.
2022
;
21
:
197
–
209
.
38.Boulet
A
, Simon
M
, Faye
G
et al.
Structure and function of the Saccharomyces cerevisiae CDC2 gene encoding the large subunit of DNA polymerase III
.
EMBO J
.
1989
;
8
:
1849
–
54
.
39.Morrison
A
, Araki
H
, Clark
AB
et al.
A third essential DNA polymerase in S. cerevisiae
.
Cell
.
1990
;
62
:
1143
–
51
.
40.Sitney
KC
, Budd
ME
, Campbell
JL
.
DNA polymerase III, a second essential DNA polymerase, is encoded by the S. cerevisiae CDC2 gene
.
Cell
.
1989
;
56
:
599
–
605
.
41.Boyce
KJ
, Cao
C
, Xue
C
et al.
A spontaneous mutation in DNA polymerase POL3 during in vitro passaging causes a hypermutator phenotype in Cryptococcus species
.
DNA Repair (Amst)
.
2020
;
86
:
102751
.
42.Darmawan
H
, Harrison
M
, Reha-Krantz
LJ
.
DNA polymerase 3'→5' exonuclease activity: different roles of the beta hairpin structure in family-B DNA polymerases
.
DNA Repair (Amst)
.
2015
;
29
:
36
–
46
.
43.Ganai
RA
, Bylund
GO
, Johansson
E
.
Switching between polymerase and exonuclease sites in DNA polymerase ε
.
Nucleic Acids Res
.
2015
;
43
:
932
–
42
.
44.Herr
AJ
, Ogawa
M
, Lawrence
NA
et al.
Mutator suppression and escape from replication error-induced extinction in yeast
.
PLoS Genet
.
2011
;
7
:
e1002282
.
45.Jin
YH
, Garg
P
, Stith
CM
et al.
The multiple biological roles of the 3'→5' exonuclease of Saccharomyces cerevisiae DNA polymerase δ require switching between the polymerase and exonuclease domains
.
Mol Cell Biol
.
2005
;
25
:
461
–
71
.
46.Jin
YH
, Obert
R
, Burgers
PM
et al.
The 3'→5' exonuclease of DNA polymerase δ can substitute for the 5' flap endonuclease Rad27/Fen1 in processing Okazaki fragments and preventing genome instability
.
Proc Natl Acad Sci USA
.
2001
;
98
:
5122
–
27
.
47.Kiktev
DA
, Dominska
M
, Zhang
T
et al.
The fidelity of DNA replication, particularly on GC-rich templates, is reduced by defects of the Fe-S cluster in DNA polymerase δ
.
Nucleic Acids Res
.
2021
;
49
:
5623
–
36
.
48.Kirchner
JM
, Tran
H
, Resnick
MA
.
A DNA polymerase ε mutant that specifically causes +1 frameshift mutations within homonucleotide runs in yeast
.
Genetics
.
2000
;
155
:
1623
–
32
.
49.Kokoska
RJ
, Stefanovic
L
, DeMai
J
et al.
Increased rates of genomic deletions generated by mutations in the yeast gene encoding DNA polymerase δ or by decreases in the cellular levels of DNA polymerase δ
.
Mol Cell Biol
.
2000
;
20
:
7490
–
504
.
50.Kokoska
RJ
, Stefanovic
L
, Tran
HT
et al.
Destabilization of yeast micro- and minisatellite DNA sequences by mutations affecting a nuclease involved in Okazaki fragment processing (rad27) and DNA polymerase δ (pol3-t)
.
Mol Cell Biol
.
1998
;
18
:
2779
–
88
.
51.Murphy
K
, Darmawan
H
, Schultz
A
et al.
A method to select for mutator DNA polymerase δs in Saccharomyces cerevisiae
.
Genome
.
2006
;
49
:
403
–
10
.
52.Nick McElhinny
SA
, Stith
CM
, Burgers
PM
et al.
Inefficient proofreading and biased error rates during inaccurate DNA synthesis by a mutant derivative of Saccharomyces cerevisiae DNA polymerase δ
.
J Biol Chem
.
2007
;
282
:
2324
–
32
.
53.Pavlov
YI
, Shcherbakova
PV
, Kunkel
TA
.
In vivo consequences of putative active site mutations in yeast DNA polymerases α, ε, δ, and ζ
.
Genetics
.
2001
;
159
:
47
–
64
.
54.Pursell
ZF
, Isoz
I
, Lundstrom
EB
et al.
Regulation of B family DNA polymerase fidelity by a conserved active site residue: characterization of M644W, M644L and M644F mutants of yeast DNA polymerase ε
.
Nucleic Acids Res
.
2007
;
35
:
3076
–
86
.
55.Shcherbakova
PV
, Noskov
VN
, Pshenichnov
MR
et al.
Base analog 6-N-hydroxylaminopurine mutagenesis in the yeast Saccharomyces cerevisiae is controlled by replicative DNA polymerases
.
Mutat Res
.
1996
;
369
:
33
–
44
.
56.Stepchenkova
EI
, Tarakhovskaya
ER
, Siebler
HM
et al.
Defect of Fe-S cluster binding by DNA polymerase δ in yeast suppresses UV-induced mutagenesis, but enhances DNA polymerase ζ—dependent spontaneous mutagenesis
.
DNA Repair (Amst)
.
2017
;
49
:
60
–
9
.
57.Williams
LN
, Herr
AJ
, Preston
BD
.
Emergence of DNA polymerase ε antimutators that escape error-induced extinction in yeast
.
Genetics
.
2013
;
193
:
751
–
70
.
58.Barbari
SR
, Beach
AK
, Markgren
JG
et al.
Enhanced polymerase activity permits efficient synthesis by cancer-associated DNA polymerase ε variants at low dNTP levels
.
Nucleic Acids Res
.
2022
;
50
:
8023
–
40
.
59.Barbari
SR
, Kane
DP
, Moore
EA
et al.
Functional analysis of cancer-associated DNA polymerase ε variants in Saccharomyces cerevisiae
.
G3
.
2018
;
8
:
1019
–
29
.
60.Castellsague
E
, Li
R
, Aligue
R
et al.
Novel POLE pathogenic germline variant in a family with multiple primary tumors results in distinct mutational signatures
.
Hum Mutat
.
2019
;
40
:
36
–
41
.
61.Daee
DL
, Mertz
TM
, Shcherbakova
PV
.
A cancer-associated DNA polymerase δ variant modeled in yeast causes a catastrophic increase in genomic instability
.
Proc Natl Acad Sci USA
.
2010
;
107
:
157
–
62
.
62.Dahl
JM
, Thomas
N
, Tracy
MA
et al.
Probing the mechanisms of two exonuclease domain mutators of DNA polymerase ε
.
Nucleic Acids Res
.
2022
;
50
:
962
–
74
.
63.Demidova
EV
, Serebriiskii
IG
, Vlasenkova
R
et al.
Correction: candidate variants in DNA replication and repair genes in early-onset renal cell carcinoma patients referred for germline testing
.
BMC Genomics
.
2023
;
24
:
388
.
64.Esteban-Jurado
C
, Gimenez-Zaragoza
D
, Munoz
J
et al.
POLE and POLD1 screening in 155 patients with multiple polyps and early-onset colorectal cancer
.
Oncotarget
.
2017
;
8
:
26732
–
43
.
65.Galati
MA
, Hodel
KP
, Gams
MS
et al.
Cancers from novel Pole-mutant mouse models provide insights into polymerase-mediated hypermutagenesis and immune checkpoint blockade
.
Cancer Res
.
2020
;
80
:
5606
–
18
.
66.Hamzaoui
N
, Alarcon
F
, Leulliot
N
et al.
Genetic, structural, and functional characterization of POLE polymerase proofreading variants allows cancer risk prediction
.
Genet Med
.
2020
;
22
:
1533
–
41
.
67.Herzog
M
, Alonso-Perez
E
, Salguero
I
et al.
Mutagenic mechanisms of cancer-associated DNA polymerase ε alleles
.
Nucleic Acids Res
.
2021
;
49
:
3919
–
31
.
68.Hodel
KP
, Sun
MJS
, Ungerleider
N
et al.
POLE mutation spectra are shaped by the mutant allele identity, Its abundance, and mismatch repair status
.
Mol Cell
.
2020
;
78
:
1166
–
77e1166
.
69.Kane
DP
, Shcherbakova
PV
.
A common cancer-associated DNA polymerase ε mutation causes an exceptionally strong mutator phenotype, indicating fidelity defects distinct from loss of proofreading
.
Cancer Res
.
2014
;
74
:
1895
–
901
.
70.Labrousse
G
, Vande Perre
P
, Parra
G
et al.
The hereditary N363K POLE exonuclease mutant extends PPAP tumor spectrum to glioblastomas by causing DNA damage and aneuploidy in addition to increased mismatch mutagenicity
.
NAR Cancer
.
2023
;
5
:
zcad011
.
71.Lee
M
, Eng
G
, Barbari
SR
et al.
Homologous recombination repair truncations predict hypermutation in microsatellite stable colorectal and endometrial tumors
.
Clin Transl Gastroenterol
.
2020
;
11
:
e00149
.
72.Li
HD
, Cuevas
I
, Zhang
M
et al.
Polymerase-mediated ultramutagenesis in mice produces diverse cancers with high mutational load
.
J Clin Invest
.
2018
;
128
:
4179
–
91
.
73.Li
HD
, Lu
C
, Zhang
H
et al.
A PoleP286R mouse model of endometrial cancer recapitulates high mutational burden and immunotherapy response
.
JCI Insight
.
2020
;
5
:
e138829
.
74.Liu
J
, Liu
Y
, Fu
J
et al.
Preliminary study on the function of the POLD1 (CDC2) EXON2 c.56G>A mutation
.
Mol Genet Genomic Med
.
2020
;
8
:
e1280
.
75.Mertz
TM
, Baranovskiy
AG
, Wang
J
et al.
Nucleotide selectivity defect and mutator phenotype conferred by a colon cancer-associated DNA polymerase δ mutation in human cells
.
Oncogene
.
2017
;
36
:
4427
–
33
.
76.Mertz
TM
, Sharma
S
, Chabes
A
et al.
Colon cancer-associated mutator DNA polymerase δ variant causes expansion of dNTP pools increasing its own infidelity
.
Proc Natl Acad Sci USA
.
2015
;
112
:
E2467
–
76
.
77.Mur
P
, Garcia-Mulero
S
, Del Valle
J
et al.
Role of POLE and POLD1 in familial cancer
.
Genet Med
.
2020
;
22
:
2089
–
100
.
78.Ostroverkhova
D
, Tyryshkin
K
, Beach
AK
et al.
DNA polymerase ε and δ variants drive mutagenesis in polypurine tracts in human tumors
.
Cell Rep
.
2024
;
43
:
113655
.
79.Pai
CC
, Heitzer
E
, Bertrand
S
et al.
Using canavanine resistance to measure mutation rates in Schizosaccharomyces pombe
.
PLoS One
.
2023
;
18
:
e0271016
.
80.Rocque
MJ
, Leipart
V
, Kumar Singh
A
et al.
Characterization of POLE c.1373A >T p.(Tyr458Phe), causing high cancer risk
.
Mol Genet Genomics
.
2023
;
298
:
555
–
66
.
81.Shinbrot
E
, Henninger
EE
, Weinhold
N
et al.
Exonuclease mutations in DNA polymerase epsilon reveal replication strand specific mutation patterns and human origins of replication
.
Genome Res
.
2014
;
24
:
1740
–
50
.
82.Shioi
S
, Shimamoto
A
, Song
Y
et al.
DNA polymerase delta Exo domain stabilizes mononucleotide microsatellites in human cells
.
DNA Repair (Amst)
.
2021
;
108
:
103216
.
83.Soriano
I
, Vazquez
E
, De Leon
N
et al.
Expression of the cancer-associated DNA polymerase ε P286R in fission yeast leads to translesion synthesis polymerase dependent hypermutation and defective DNA replication
.
PLoS Genet
.
2021
;
17
:
e1009526
.
84.Weber
CAM
, Kronke
N
, Volk
V
et al.
Rare germline variants in POLE and POLD1 encoding the catalytic subunits of DNA polymerases ε and δ in glioma families
.
Acta Neuropathol Commun
.
2023
;
11
:
184
.
85.Xing
X
, Kane
DP
, Bulock
CR
et al.
A recurrent cancer-associated substitution in DNA polymerase ε produces a hyperactive enzyme
.
Nat Commun
.
2019
;
10
:
374
.
© The Author(s) 2025. Published by Oxford University Press.
This is an Open Access article distributed under the terms of the Creative Commons Attribution License (
https://creativecommons.org/licenses/by/4.0/), which permits unrestricted reuse, distribution, and reproduction in any medium, provided the original work is properly cited.







{kind=link}
{kind=link}
{kind=link}