





Abstract
Protein–protein interaction (PPI) databases with structural information are useful to investigate biological functions at both systematic and atomic levels. However, most existing PPI databases only curate binary interactome. From the perspective of the display and function of PPI, as well as the structural binding interface, the related database and resources are summarized. We developed a database extension, named mPPI, for PPI structural visualization. Comparing with the existing structural interactomes that curate resolved PPI conformation in pairs, mPPI can visualize target protein and its multiple interactors simultaneously, which facilitates multi-target drug discovery and structure prediction of protein macro-complexes. By employing a protein–protein docking algorithm, mPPI largely extends the coverage of structural interactome from experimentally resolved complexes. mPPI is designed to be a customizable and convenient plugin for PPI databases. It possesses wide potential applications for various PPI databases, and it has been used for a neurodegenerative disease–related PPI database as demonstration. Scripts and implementation guidelines of mPPI are documented at the database tool website.
Database URL http://bis.zju.edu.cn/mppi/
Introduction
Protein–protein interaction (PPI) networks provide valuable information to understand cellular mechanisms. PPI networks in the node-and-edge presentation are helpful for a global comprehension of biological processes. To discover details on the mechanisms of how proteins execute their functions and how disease-related mutations disturb cellular homeostasis, it is helpful to import structural details of protein interactions to these abstract networks (1).
However, currently, most PPI databases are not equipped with interaction structures, resulting in an urgent need of quaternary coverage for these databases to boost biomedical studies. These databases curate various interactomes with diverse time, disease and cell-type specificities; thus, a widely applicable database extension for structural presentation is ideal.
The situations vary in the availability and the visualization technology of the PPI databases established to show the structural details of protein interactions. The datasets turned outdated after these platforms were being released for years. Besides, experimental data were insufficient to support protein structure interaction, so that some calculation methods are required. The few existing PPI databases with structural annotation, such as Interactome3D (2), INstruct (3) and Interactome INSIDER (4), have been developed to aid biological discoveries by visualizing protein interaction structures in pairs. However, the pairwise presentation has its limitations as it only shows the target protein and one of its interactors at one time. First, protein generally employs multiple interactors into complexes to perform functions. For example, it was verified that protein ASC has two distinct binding sites, which interact with protein NLRP3 and POP1 simultaneously to trigger immune response (5). It is helpful to examine the structural compatibility of multiple interactors and their potential binding structure before experimental verification. Second, recently, the multi-target pharmacology became increasingly auspicious to cure complex diseases. This so-called polypharmacology suggests that effective drugs can be developed by interfering with multiple receptors in the disease networks (6, 7). One efficient way to identify a group of proteins that could be potentially targeted together is to project their binding interfaces in one scene and discover their overlaps.
Besides, compared to the whole interactome, there is only a tiny fraction of experimentally resolved protein complexes, requiring in silico predictions to accommodate the increasing need of a fully decoded structural interactome (2, 8, 9). Computational methods, e.g. homology modeling (10), machine learning (4) and protein–protein docking (11), have been developed to infer protein interaction structures. Homology-based algorithms rely on structural templates and could only propose a limited number of complex structures. It is counted that cocrystal structures and homology models together cover ∼6% of all known interactions (4). Machine learning algorithms work on any pair of protein structures but only predict their binding sites, ignoring the complete structure that is also essential for biological interpretation. Therefore, docking algorithms that calculate the overall interaction conformation on the whole interactome is a reasonable option.
Here, we propose mPPI, a database extension to display structural interactome in a one-to-many manner. By employing a protein–protein docking algorithm and three-dimensional (3D) molecule visualization platform, mPPI largely extended the presentation scope from resolved protein complexes, in the manner that projects the interaction structures of the target protein and all its interactors simultaneously. mPPI can benefit database developers to conveniently integrate a structural visualization platform into their databases. The resulting structural interactomes with the multiway presentation will further aid biomedical experts in their research.
Methods
Methods for reviewing existing PPI databases
Multiple sources were integrated to search for entries released after 2010 in which the category or the description includes ‘protein–protein interaction database’, ‘structure’ or any terms related to binding interfaces. The retrieved results were then manually curated, tagged and deduplicated. Each database is tested for availability and the existence of binding interface visualization, through actual access on the modern web browsers. The results are sorted according to availability, visualization capabilities and the year of last update.
Construction of mPPI
There are two modules in mPPI: ‘dock’ and ‘viz’ (Figure 1). When database developers are integrating this extension into their databases, the ‘dock’ module will extract their binary PPI information, download corresponding structural files and calculate their pairwise interaction conformation; then, the ‘viz’ module will build a visualization platform into the database, where previous calculations will be displayed in a one-to-many manner.
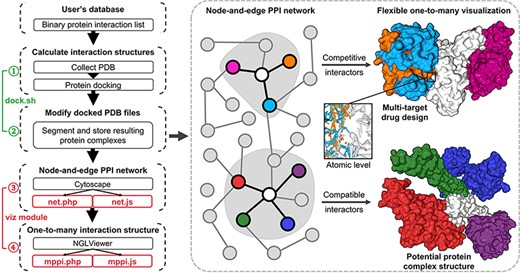
Illustration of mPPI. The left part is the workflow of mPPI. After applying mPPI, the user’s database will acquire visualization functions demonstrated in the dashed box. In the dashed box, the left part is the 2D network representing binary protein–protein interaction from the user’s database. Any node in the network could be chosen to visualize its multiple interactors. The one-to-many structural presentation is highly flexible that the visibility, scale, angle and presentation style of each interactor can be adjusted for the most intuitive perspective. Two examples are demonstrated on the right side of the dashed box, where the colors correspond between the protein nodes and the 3D structures. Structurally compatible and competitive interactors are selected for visualization to assist protein macro-complex structure prediction and multi-target drug design. Atomic-level presentation is supported to further aid drug design and mechanism discovery, as shown in the middle of the dashed box.
Predict pairwise interaction
This step will extract binary interaction information in users’ databases and process it into raw materials for structural visualization. The PPI network documented in the user’s database will be transformed into a binary interaction list (multiple lines with two protein IDs in a line), in accord with their pairwise interaction information. mPPI will download their corresponding PDB files. For each pair of interacting proteins, mPPI will then employ ZDOCK 3.0.2 (12) to calculate their potential complex structure with the highest docking score. The interaction structures and docking scores will be stored for the next steps.
Modify docked PDBs
The raw materials are processed so that they can then be visualized by the ‘viz’ module. It will segment every docked protein complexes into two PDB files as two interactors and store the resulting files into folders in a specific manner (each folder corresponds to a protein node in the interactome, which contains docked PDB files of the protein node and all its interactors), so that the visibility, scale, angle and presentation style of each interactor can be adjusted separately. The previous step ‘Predict pairwise interaction’ and this step are integrated into one script ‘dock.sh’.
Construct abstract PPI network
This step will integrate a node-and-edge network into users’ database. A JavaScript library Cytoscape (13) is employed to display 2D protein interaction networks. Two scripts ‘net.js’ and ‘net.php’ are created to present a 2D interactome based on the binary interaction list.
Visualize one-to-many interactions
This step is the main part of our tool, which represents the structures in a network perspective. The tool employs NGL viewer v2.0.0-dev.39 (14) as the backbone library to visualize macromolecule structures. Two scripts ‘mppi.js’ and ‘mppi.php’ are in charge of this step. Given the pairwise interaction structures, this step will present the docked structures of the target protein and all its interactors, where the visibility, scale, angle and presentation style of all components can be freely adjusted.
Calculate compatibilities of protein ligands
Apart from visualizing the structural relationships between protein ligands, another major contribution of mPPI is to directly calculate their compatibilities. For each pair of ligands binding to the same receptor, the system will discover the closest two atoms from each ligand. If the Euclidean distance of the two atoms is above a threshold (2 Å), the two ligands are considered compatible, otherwise noted competitive. When users operate on the selections and unselections of protein ligands, the other ligands compatible or competitive to the current selected ligands will be tagged accordingly on the visualization system.
More methodological descriptions, format requirements, step-by-step user guidelines and example applications can be referred to the online document.
Results
Review of the PPI databases selected by features
In this study, some features were selected to summarize the situation of mainstream PPI databases, especially from the perspective of 3D visualization of binding interface details.
Through the search in multiple sources, 223 duplicate entries were obtained. The duplication was because most indexed entries were from literature mining, which has not excluded the submission of database updates. After manual curation and deduplication, 41 databases more relevant to this research established after 2010 were selected from the entries (Table 1).
Existing database and resources of PPI related to binding interfaces
Existing database and resources of PPI related to binding interfaces
These database entries are annotated with the last update time, the application of binding interface visualization, the visualization method implemented and availability. Their subjects include multispecies, humans and plants. Eight of the 41 databases applied the structural visualization of the binding interface (2, 4, 15–20). Different plugins were implemented for visualization, such as PyMOL, JSmol, 3Dmol and Flash-player-based.
KBDOCK is established to define the protein binding sites and perform spatial clustering on them to achieve knowledge-based protein docking (15). In the 3D system, for each query domain, a non-redundant list of domain–domain interactions (DDIs) and a Jmol view of the DDIs was shown.
3did is a knowledge-based catalog of PPIs with high-resolution 3D structures (16). It provides critical molecular details explaining the emergence of PPIs and offers an overview of the similarity in the interactions of different proteins in the same family. Since the Flash player reached the end of life on 1 January 2021, the 3did’s visualization based on it can no longer work properly in most modern browsers.
Piface is a clustered protein–protein interface database collecting non-redundant unique interface structures from PDB (17). Search functions by interfaces and by domains were implemented. The corresponding domains to the queried interface and the interface similar to it can be retrieved and visualized by JSmol.
HotRegion provides information on the hot region of the interface by predicted hot spot residues, the structural characteristics of these interface residues, as well as the 3D visualization of the interface and the interaction between hot regions (18).
InterEvol database is for the combined structural and evolutionary analysis of protein complex interfaces (19). It systematically analyzed the PDB chains’ interfaces and complexes and clustered them to capture the characteristics. The visualization was presented by the PyMOL plugin.
Seventeen databases have been updated since the year 2019, two of which belong to those equipped with the binding interface structure visualization (2, 4, 21–35). It can be inferred that the lack of structural calculation and visualization of the binding interface datasets is one of the difficulties in timely updates. The PPI databases without binding interface visualization or a recent update were also collected (36–47). Six databases are currently inaccessible as websites (20, 48–52), including PICCOLO, a relational database on the details of PPI structures (20). Except for the BIND database that only provides data download access (52), these databases either have expired domain names or are under maintenance.
During the investigation, it was found that limited experimental data or protein dimers were applied by some databases to visualize the structure of the binding interface. These databases contain neither enough protein pairwise interaction structures nor calculation and visualization of one-to-many protein interactions. It is assumed that a flexible workflow can help PPI databases with limited binary interaction information to establish docking calculation and detailed structural visualization in a one-to-many way.
mPPI as a PPI database extension
mPPI is designed as a customizable and convenient database extension. It has wide potential applications to existing databases. Here, we demonstrate its successful utility in a PPI database focusing on neurodegenerative diseases (ND) (http://bis.zju.edu.cn/ndatlas/). We select a sub-interactome centered at protein JUN to illustrate the utility of mPPI and how mPPI will boost biological discovery and drug design (Figure 2).
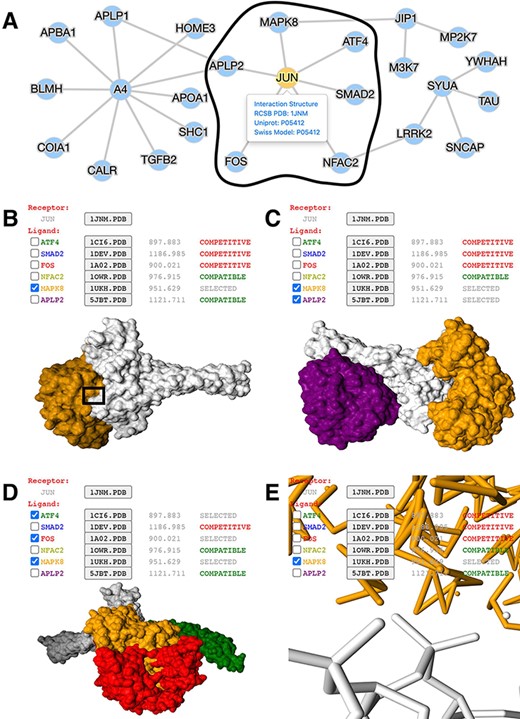
Application of mPPI in a disease-related PPI database. (A) PPI network in node-and-edge representation. This example network is a sub-interactome containing 25 proteins related to ND (http://bis.zju.edu.cn/ndatlas/). The black circle stressed out a sub-network center at protein JUN that will be structurally displayed in Fig. 2B–E. (B) Presentation style and logistics of multiway structural interactions in mPPI. When selecting proteins to interact (in this example a protein MAPK8), the selected proteins will be tagged ‘SELECTED’, and all other proteins that are structurally compatible to them will be tagged ‘COMPATIBLE’, while other competitive ligands will be tagged ‘COMPETITIVE’, in the last column of each protein row. The checking and unchecking of each protein will change all the compatibility tags accordingly. (C) Illustration of one-to-many structural interaction. The center protein is JUN (white), and we select two compatible interactors MAPK8 (orange), APLP2 (purple) to visualize. (D) Another illustration of one-to-many structural interaction. The center protein is JUN (white), and interactors ATF4 (green), FOS (red) and MAPK8 (orange) are competitive. (E) PPI conformation at atomic resolution. The partial image of structural interaction stressed out in black box in Fig. 2B intuitively displays the binding interface of JUN and MAPK8. For each protein ligands, the docked structures can be downloaded for users’ further analysis by clicking the ‘.PDB’ button behind each protein name, and the docking scores (representing the tightness of the interaction calculated by ZDOCK) are shown behind each ‘.PDB’ button.
The node-and-edge network overviews the topology of the interactions (Figure 2A). By clicking the target protein of interest, in the case of protein JUN, all its interactors’ conformations and docking scores can be freely displayed. Upon selection of protein ligands, the compatibility of other interactors will be clearly indicated. Users can select either compatible or competitive ligands of interest for further studies, as demonstrated in Figure 2B.
Figure 2C presents the structure of protein JUN and two interactors MAPK8 and APLP2. These interactors have distinct interfaces to JUN with nonoverlapping conformation, which indicates these compatible proteins will possibly bind simultaneously and form a macro-complex. Also, the design of inhibitors to prevent JUN-triggered neurodegeneration will require exhaustive consideration of these four discrete binding sites (53). Moreover, this suggests that JUN evolves slower and performs more critical functions in ND, compared to other ND-related hub proteins with fewer interfaces (54).
When the other three interactors MAPK8, FOS and ATF4 bind to JUN, there is a large area of overlapping interface and conformation (Figure 2D). This indicates that their shared interface is promiscuous, with fewer non-synonymous single-nucleotide variations and limited resilience to disease-causing mutations (55). Besides, their interactions are competitive with a highly overlapping interface, which forecasts potential drug side effects and off-targets (1). However, in the concept of polypharmacology, their overlap that is clearly shown by mPPI can be targeted together and are promising drug binding sites to cure complex diseases.
Besides, mPPI provides structural details at an atomic level. Figure 2E displays the selected interaction area of the atoms and dynamic bonds of JUN and MAPK8, which brings intuitions in predicting concrete cellular mechanisms and deciding drug binding sites.
Discussion
As a convenient and customizable plugin, mPPI will help database developers to visualize structural interactions in their diverse databases, which largely boosts the popularization of structurally resolved PPI databases in the future. Databases enhanced by this tool will then largely benefit biomedical experts and structural biologists, as mPPI also innovates on its presentation style that clearly visualizes the structural relationships of target protein’s multiple interactors, therefore offers unprecedented perspective to cellular mechanism discovery and drug design. As for now, mPPI has been successfully embedded into a PPI database focusing on neurodegeneration and has already shown novelty and potential in biomedical studies, which promises its value to the general audience above.
Considering the lack of quaternary coverage in existing PPI databases, the main goal of mPPI is set to the popularization of structurally resolved PPI databases, with innovations in its presentation style and functionalities. As the development of structural interactome becomes increasingly prevalent, the research focus will extend from its creation to validation and refinement. mPPI also has huge potentials in these fields given proper adjustments. The following possible research directions will be considered in the next version of mPPI.
Online query of small structural interactomes
The first-hand users of mPPI are database developers, so it was inevitable for this tool to implement local docking algorithms and web scripts in JavaScript and PHP. Although researchers interested in specific interaction groups may use a more coherent workflow, mPPI still provides a flexible interface to adapt to future development.
mPPI can be adjusted into a platform for online queries of small structural interactomes. Experts can upload their PPI network of interest on the website and be responded with a visualization of structural interactome after online calculation. In this scenario, an online docking server, e.g. HDOCK (56), may replace local docking scripts that mPPI originally implemented.
PPI network imputation and refinement
In the current version, the PPI networks are provided by the database developers, which are already validated by the developers and within a specific biomedical context, so mPPI is designed to present its structural network consistent with the original binary interactome. However, there are still urgent needs for the prediction and validation of protein interactions. The system can also be tuned for the imputation and validation of protein interaction networks. Given an interactome, the system will predict its corresponding multiway protein complexes. Based on the predicted contact information, the system can propose the likelihood of each protein interaction. The system can be integrated into the mentioned online query platform to improve its utility.
Refinement of macro-complex structure
Aiming to provide a user-friendly platform for the viewers of mPPI-implemented databases, the tool is set to visualize the interaction structures with the highest docking scores. Although presenting lower scored conformations into the visualization platform may confuse the viewers, these alternative structures may be useful for further refinement of macro-complex structures. Under this setting, mPPI can be altered into an algorithm for macro-complex structure prediction in high resolution. The algorithm can integrate rich sequence information from meta-genome sequencing for further refinement. The algorithm can also be improved using the insights from multidomain protein structure prediction (57). The algorithm can be integrated into the online query platform that we previously mentioned.
Funding
National Key Research and Development Program of China (2016YFA0501704, 2018YFC0310600); National Natural Sciences Foundation of China (31571366, 31771477, 32070677); Jiangsu Collaborative Innovation Center for Modern Crop Production and the Fundamental Research Funds for the Central Universities.
Conflict of interest.
None declared.
Author biographies
Yekai Zhou is a research assistant in Ming Chen’s laboratory in Zhejiang University. His research focuses on Structural Bioinformatics and Medical Artificial Intelligence.
Hongjun Chen is a PhD candidate in Ming Chen’s laboratory in Zhejiang University. His research focuses on protein–protein interaction networks and disease-involved network modules.
Sida Li is a PhD candidate in Ming Chen’s laboratory in Zhejiang University. His research focuses on single-cell multiomics integration and biomedical text mining.
Ming Chen is a full professor in the Department of Bioinformatics, College of Life Sciences, Zhejiang University. He is the president of Bioinformatics Society of Zhejiang Province, China. His current research focuses on establishing useful bioinformatics tools and platforms to help biologists browse and analyze massive biological datasets.
Contributions
Y.Z. developed the tool and wrote the manuscript draft. H.C. and M.C. evaluated tool performances. H.C and S.L. contributed to the review of existing PPI databases. M.C. supervised the project. All authors contributed to and approved the final manuscript.
Key points
The established database and resources related to the structural binding interface were summarized and reviewed.
The proposed tool mPPI provides a structure visualization platform for online PPI databases.
mPPI projects the overall conformations of multiway protein interactions.
mPPI helps multi-target drug design and protein complex structure prediction.
Code and guidelines are available at http://bis.zju.edu.cn/mppi/.

{kind=link}
{kind=link}