





Abstract
Gramene is a comparative information resource for plants that integrates data across diverse data domains. In this article, we describe the development of a quantitative trait loci (QTL) database and illustrate how it can be used to facilitate both the forward and reverse genetics research. The QTL database contains the largest online collection of rice QTL data in the world. Using flanking markers as anchors, QTLs originally reported on individual genetic maps have been systematically aligned to the rice sequence where they can be searched as standard genomic features. Researchers can determine whether a QTL co-localizes with other QTLs detected in independent experiments and can combine data from multiple studies to improve the resolution of a QTL position. Candidate genes falling within a QTL interval can be identified and their relationship to particular phenotypes can be inferred based on functional annotations provided by ontology terms. Mutations identified in functional genomics populations and association mapping panels can be aligned with QTL regions to facilitate fine mapping and validation of gene–phenotype associations. By assembling and integrating diverse types of data and information across species and levels of biological complexity, the QTL database enhances the potential to understand and utilize QTL information in biological research.
Introduction
Gramene is a comparative genome database for plants that integrates information about genetic and physical maps, sequences, markers, germplasm resources, genes, proteins, pathways and phenotypes (1–3). Users can browse or query the database to discover relationships between genes and phenotypes of interest. They can draw on data from multiple plant species to compare and contrast the characteristics of genes, genomes, pathways and phenotypes.
This article describes the annotation of quantitative trait loci (QTL) within Gramene and illustrates the way this resource can be used to identify genes and regulatory sequences underlying QTLs using both the forward and reverse genetics approaches. The QTL database is a one of a kind resource; it contains the largest online collection of rice QTL data in the world and serves as a repository for the international rice research community. It facilitates the investigation of QTLs in co-linear regions in other cereals and enables researchers to identify sequences and QTLs associated with similar traits or phenotypes across a wide range of plant species through the use of controlled vocabularies. By providing an integrated set of tools, it offers plant biologists and geneticists a way of exploring the relationship between genome variation and complex forms of phenotypic variation (4–7).
In Gramene, a QTL is identified as a region of the genome that is predicted to contain a gene or genes associated with a specific trait. QTL mapping involves the analysis of a population(s) where individual plants, lines or families within the population have been characterized for a set of well-distributed molecular marker polymorphisms as well as for one or more quantitative traits. A QTL is declared when there is a statistical association between the segregation of a molecular polymorphism(s) and a measurable phenotype, using the individual segregants within the population as replicates. The phenotype of interest may be a feature of the whole plant, an organ or a tissue, or it may be characterized as a feature associated with the DNA, RNA or protein. The objective of QTL analysis is to identify the position and relative importance of genetic factors that collectively determine a trait or a phenotype of interest (6,8).
QTL curation contributes to the functional annotation of the rice genome. QTLs, in effect, function as genomic placeholders; they flag positions in the genome that harbor genes underlying traits of interest. QTL mapping has been particularly relevant to the agricultural community because it provides a way of genetically dissecting quantitative variation found in naturally occurring germplasm resources and offers insight into the linkage and epistatic relationships among genes and QTLs controlling diverse traits of interest. Furthermore, plant breeders are able to make direct use of QTL results for marker-assisted selection in breeding programs. QTL analysis is also used by molecular geneticists as a first step in map-based cloning studies and it provides quantitative geneticists and evolutionary biologists with a global view of gene network architecture, allowing them to identify key rate-limiting steps associated with quantitative variation (5,7). By reducing the search space, QTL information makes it easier to identify individual genes underlying quantitative traits and provides global information about the location and relative importance of each genetic factor (4,9). QTLs are highly informative because they have integrative power to connect diverse domains of information in the plant biology. This is vital to understanding the biology of complex traits and serves a critical function in database curation and design.
One of the most pressing reasons to curate QTLs is that, in recent years, major resources have been invested in plant and animal QTL research worldwide; this has generated a large volume of QTL information in the published literature. The value of an information resource or central repository that can assemble and integrate QTL information across species and levels of biological complexity is underscored by the fact that the information is reported in different formats, and the data are highly heterogeneous and often fragmentary. Public databases have a responsibility to harness as much information as possible and organize it into useful online resources for use by diverse research communities (8,10,11).
A majority of the rice QTLs published between 1994 and 2007 have been curated from the literature and are currently available in the Gramene database. This was accomplished by extracting phenotypic information from highly heterogeneous textual descriptions and codifying it, using structured vocabularies and ontologies (2,10,11). Meanwhile, the thousands of diverse molecular markers that were used to map QTLs in hundreds of different populations were associated to sequences that could be aligned to the rice genome. As a result, the complex relationships between phenotypes and genotypes encoded by each QTL have now been organized into a network of properties and information in the context of a genome that is searchable and retrievable within the Gramene database.
Materials and methods
QTL data sources
Literature
To identify publications reporting rice QTLs, Gramene curators queried public library databases including PubMed, Agricola, BIOSIS Previews and CAB abstracts using keywords such as ‘Oryza/rice and quantitative trait loci/QTL’. After screening search results for relevance, each publication to be curated was assigned a unique reference ID and all curated QTLs were referenced to the original paper. All in all, between 1994 and 2007, thousands of research papers were scanned for QTL data and deposited in the Gramene literature database. The QTL data extracted from the literature included QTL names, symbols, traits, associated co-localized and neighboring markers, parental strains, types of crosses and other pertinent information.
Information to be entered into the database was extracted based on a set of priorities. The top priorities included information required to establish the genome position of a QTL, and information describing the trait or phenotype associated with each QTL. Trait descriptions were mapped to controlled vocabularies including the Trait Ontology (TO), Plant Ontology (PO) and Plant Growth Stage Ontology (GSO) (10–12). A detailed standard operating procedure (SOP) for QTL curation designed for curators and researchers who are at the initial stages of setting up a QTL database is available at http://ascus.plbr.cornell.edu/∼gramene/qtl_ms/sop/.
Integration of QTLs from MaizeGDB and GrainGenes
Collaborating with MaizeGDB (http://www.maizegdb.org/) and GrainGenes (http://wheat.pw.usda.gov/GG2/index.shtml), QTLs from maize, wheat, barley and oats, originally curated by those databases (13,14) have been integrated into Gramene. The schemas of those two databases and Gramene were compared, and the correspondences between data entries were identified. Additional curation was performed as necessary to meet standards set by Gramene.
Defining a single QTL in Gramene
Each experimentally defined QTL is treated as an independent entity in Gramene. This is designed to reduce the number of tracking/merging issues confronted by the curators and it ensures that QTLs are treated as statistical hypotheses rather than as confirmed genetic entities. In this way, if QTLs were reported in two different papers (even from the same group and with similar genetic materials), or if two QTLs were detected for the same trait in the same location but based on experiments in different years or locations, or if QTLs were identified by different QTL analysis methods (e.g. one-way Analysis of variance (ANOVA) versus Composite Interval Mapping), each QTL citation is curated as an independent entity, even where the authors reported them as a single QTL. This allows database users to weigh the evidence for the existence of a QTL based on the data; they can assess the number of times a putative QTL is reported in a similar location in different experiments. This approach avoids the assumption that a single genetic factor is inevitably responsible for a particular phenotype, leaving room for confirmation once a specific gene or functional allele(s) has been cloned and characterized.
QTL nomenclature
Each QTL is assigned a unique accession identifier in Gramene, e.g. ‘NQA001’. In this case, the first character (N) serves as a tracking mechanism to identify the curator who handled the information; Q stands for QTL; the third character (A) indicates the reference; and the following three digits are used to distinguish different QTLs from the same publication. This nomenclature is useful because it provides a mechanism for tracking all QTL/sets of QTL managed by a particular curator or all QTLs that correspond to a particular study or paper. This system also addresses management issues within the schema and most importantly provides a mechanism that allows users to query and download QTLs from the database.
Curation of genetic information in a QTL study
Curation of genetic information associated with QTLs is done in two stages in Gramene, namely QTL map data are first compiled and then QTL intervals are assigned, as outlined below.
QTL map data
Information about a QTL map is filled out by a curator, who first verifies whether the same population and genetic map information may have been used in a prior dataset that has already been curated in Gramene. If this information is already present in the database, it must be checked to see whether there were any modifications to the Map Set between studies. To determine this, the curator must compare the information in both studies to determine whether they are identical in terms of population size and structure, male and female parental identity, type and number of markers and the corresponding map display. If the Map Set is identical to a previously curated Map Set, the data already in the database are used as the reference map. If the details of the Map Set are significantly different, a new data file is created based on the current paper. Discrete positions for all markers on the map must be entered into a map file. In many cases, a published paper may not report the exact map position/interval distances for all markers. In such cases the curator contacts the corresponding or first author of the paper to obtain the required information.
Assignment of QTL intervals
The second important type of information extracted from a published paper is the QTL interval. This interval represents the search space associated with the QTL and within which any gene underlying the QTL is expected to reside. If the QTL interval has been clearly determined (i.e. the paper specifies that the QTL extends from marker a to b, or from cM x to cM y on the map, the positions of the interval for the QTL will be used as the feature start (upper) and feature stop (lower) positions along the chromosome. If the QTL interval was not clearly delineated in the paper, but a ‘peak marker’ was identified, the positions of the two most closely linked flanking markers (on either side of the QTL peak) are used as the start and stop positions for that QTL (the upper marker position as start position, and lower marker as stop position). If only a single marker is mentioned in association with a QTL (i.e. results from single point analysis), that marker's position alone is used as both the start and stop position for the QTL. The linkage group or chromosome number to which a particular QTL is mapped is also included in the data table.
Anchoring of genetic intervals to genomic sequence
The mapping of rice QTLs to genomic positions has been standardized by Gramene as follows. QTLs are defined by flanking or closely linked molecular markers identified from the original published QTL map. When the markers are restriction fragment length polymorphisms (RFLPs), their nucleotide sequence is obtained and BLAT (BLAST-like alignment tool) (15) is performed to obtain the genome position of that RFLP marker. If the markers are microsatellites, or simple sequence repeats (SSRs), the primer sequences are obtained and e-PCR is performed to determine the marker positions on the sequenced genome. In cases where a critical RFLP or SSR marker cannot be mapped unequivocally to the rice genome, but one or more markers in the interval associated with a particular QTL can be mapped to the expected genomic region, that marker is used to anchor the QTL to the genome and its position defines the location of the QTL on the genome map.
Curation of phenotypic information in a QTL study
The curators at Gramene have established protocols for extracting and encoding the highly heterogeneous phenotypic information associated with QTL studies using a combination of ontologies, evidence codes and free text assignments. These protocols are outlined below.
Development of vocabularies to describe plant phenotype
Gramene curators are developers and collaborators of the Plant Ontology Consortium (POC) (http://www.plantontology.org/). The POC develops and maintains controlled vocabularies or ontologies for Plant Ontology (PO) and plant growth and development (GSO) for the purpose of annotation (10,11,16). Additional vocabularies/ontologies used in Gramene, such as the Environmental Ontology (EO) and Trait Ontology (TO), were developed in-house. The TO (12) is developed based on phenotypic assays and vocabulary used widely in the agronomy and plant breeding communities. These ontologies are under continuous maintenance with new terms being actively added, as and when curators or users request them for annotation of new phenotypic traits.
Association to ontology terms
Each QTL is associated with a trait name corresponding to a term in the TO. Trait symbols are derived from the trait name, e.g. plant height carries the symbol PTHT, and are used to display the QTL position on a linkage group. The QTL is displayed as a feature on a map in the comparative map viewer, CMap (http://gmod.org/wiki/CMap) and in the rice genome browser. There are nine trait categories corresponding to agronomic or plant breeding classifications, and they include Yield, Grain Quality, Biotic Stress, Abiotic Stress, Sterility/Fertility, Vigor, Anatomy, Development and Biochemistry.
The published symbol for the QTL corresponds to the name or symbol described by the author in the original publication. Each QTL is also associated with an anatomical portion of the plant and a specific growth or developmental stage corresponding to the organ or tissue and time of development in which the trait was evaluated. In Gramene, terms from PO (10) and GSO (11) are used to describe these anatomical features and developmental stages. In addition, the environmental conditions and any supplemental treatment(s) used to determine the phenotype are recorded using terms from the EO.
Use of evidence codes
In Gramene, all QTL annotations are supported by the use of evidence codes. These codes indicate what data are available in the literature to support a variety of inferences, including ‘Inferred by association of genotype from phenotype’ (IAGP), ‘Inferred by curator’ (IC), ‘Starting material’ (SM). In addition, to the ones developed by the Gene Ontology database, a few evidence codes have been developed in-house. Evidence codes are also used to denote specific associations to ontology terms based on information in the published paper describing the QTL.
Database schema
The schema developed for the QTL database and all QTL data are downloadable from the Gramene ftp site: ftp://ftp.gramene.org/pub/gramene/CURRENT_RELEASE/data/database_dump/mysql-dumps.
Results
Phenotypes associated with QTLs are evaluated using specific genetic materials that provide contrasting phenotypic states. Historically, bi-parental populations derived from controlled crosses were used for identifying QTLs, but increasingly QTLs are being identified via association or linkage disequilibrium mapping (17). In any QTL study, specific traits or phenotypic features are assayed under a defined set of environmental conditions. A genetic study describing the relationship between phenotype and genotype will embody a series of interactions among loci and alleles in the genetic background, as well as interactions between genotype and the environment(s) in which the population is assayed. The curator tackles the complex problem of describing the relationship among each different data element (gene, allele, genetic population, phenotype and environment) and linking each element to all other entries in the database using a combination of bioinformatic tools, ontologies and free text.
QTL data acquisition and significance
The first plant QTL paper was published in 1988 (18), and since that time thousands of plant QTL studies have been published, including 617 papers reporting rice QTLs, 454 on wheat and 364 on maize. Figure 1 summarizes the number of QTL papers cited for rice, wheat, maize, tomato and Arabidopsis in the four major reference databases over the last 21 years. In Gramene, more than 230 papers have been curated for rice. This number is only 38% of the total number of rice publications because the Gramene curators impose certain requirements on what is to be included in the database. Their criteria include: (i) availability of mapset information; (ii) use of sequence-based markers for anchoring QTL to the genome (Amplified fragment length polymorphism (AFLP) and Random amplification of polymorphic DNA (RAPD) markers, for example, cannot be aligned); (iii) availability of published information in English. After eliminating papers that did not meet these criteria, the number of qualifying rice QTL papers was cut to just under 40%.
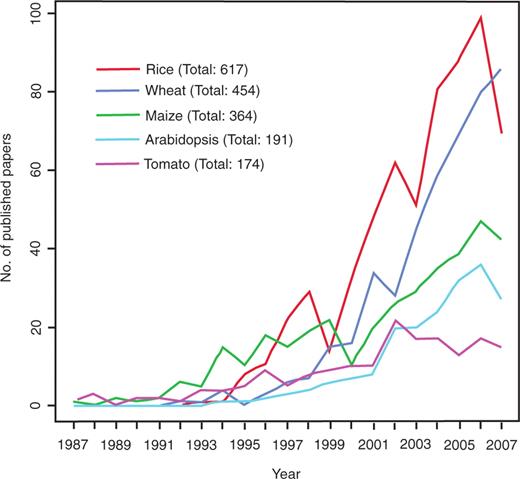
Number of published QTL papers for rice, wheat, maize, Arabidopsis and tomato between 1987 and 2007. Graph showing the steady increase in publications reporting QTLs in five major plant species between 1987 and 2007 based on nonredundant data from four publicly available literature databases, PubMed, Agricola, CAB Abstracts and BIOSIS Previews.
QTL statistics in Gramene Build 28
The QTL module in Gramene contributes to the functional annotation of the rice genome, a process that involves continuous layering of information onto the sequenced genome, by delineating the genome into thousands of specific regions that have a high probability of containing genes controlling quantitative traits. Active curation of QTLs in the Gramene database began in the year 2003 and since then more than 11 000 QTLs have been curated belonging to rice, pearl millet, foxtail millet, maize, wheat, barley and oat (Table 1). The number of QTLs curated for rice is much higher than those from other species, reflecting Gramene's priority to focus on rice as the first sequenced crop genome and corresponding to the higher rate of QTL identification in rice. The number of rice QTLs that have been projected on to the 12 rice chromosomes are given in Table 2. The rapid increase in the number of published QTL papers (Figure 1) requires a consistent effort to source this information, integrate it into the database and keep it current. The QTL curation is labor intensive and there are few experts prepared to handle this aspect of data curation, so information is extracted in phases and new methods are being developed to help automate this procedure. Extensive quality control protocols have been developed and put into practice to ensure accuracy of the curated data.
Summary of QTL and associated features in the Gramene database for 10 cereal species
| Updated based on build 28. | Rice | Maize | Wheat | Tetraploid wheat | Oat | Barley | Sorghum | Pearl millet | Foxtail millet | Wild rice | Total |
|---|---|---|---|---|---|---|---|---|---|---|---|
| QTLs | 8646 | 1747 | 23 | 8 | 375 | 299 | 136 | 284 | 65 | 41 | 11624 |
| TO: traitsa | 237 | 77 | 10 | 3 | 7 | 30 | 19 | 27 | 2 | 10 | 332 |
| TO: trait categoriesb | 9 | 8 | 3 | 2 | 5 | 7 | 5 | 6 | 1 | 5 | 9 |
| PO: structurec | 38 | 19 | 5 | 2 | 6 | 8 | 7 | 15 | 2 | 5 | 48 |
| PO: growth-staged | 19 | 9 | 6 | 2 | 5 | 5 | 7 | 7 | 2 | 5 | 20 |
| Map setse | 89 | 8 | 9 | 2 | 1 | 8 | 2 | 5 | 1 | 1 | 126 |
| Parental germplasmf | 91 | 4 | 4 | 2 | 2 | 7 | 3 | 12 | 2 | 2 | 108 |
| Co-localized markersg | 30950 | 3615 | 73 | 14 | 888 | 335 | 334 | 1031 | 87 | 42 | 37369 |
| Neighboring markersh | 16422 | 3120 | 37 | 14 | 561 | 258 | 558 | 535 | 122 | 74 | 21671 |
| Curated papers | 246 | 56 | 11 | 2 | 1 | 9 | 2 | 6 | 1 | 1 | 335 |
| Updated based on build 28. | Rice | Maize | Wheat | Tetraploid wheat | Oat | Barley | Sorghum | Pearl millet | Foxtail millet | Wild rice | Total |
|---|---|---|---|---|---|---|---|---|---|---|---|
| QTLs | 8646 | 1747 | 23 | 8 | 375 | 299 | 136 | 284 | 65 | 41 | 11624 |
| TO: traitsa | 237 | 77 | 10 | 3 | 7 | 30 | 19 | 27 | 2 | 10 | 332 |
| TO: trait categoriesb | 9 | 8 | 3 | 2 | 5 | 7 | 5 | 6 | 1 | 5 | 9 |
| PO: structurec | 38 | 19 | 5 | 2 | 6 | 8 | 7 | 15 | 2 | 5 | 48 |
| PO: growth-staged | 19 | 9 | 6 | 2 | 5 | 5 | 7 | 7 | 2 | 5 | 20 |
| Map setse | 89 | 8 | 9 | 2 | 1 | 8 | 2 | 5 | 1 | 1 | 126 |
| Parental germplasmf | 91 | 4 | 4 | 2 | 2 | 7 | 3 | 12 | 2 | 2 | 108 |
| Co-localized markersg | 30950 | 3615 | 73 | 14 | 888 | 335 | 334 | 1031 | 87 | 42 | 37369 |
| Neighboring markersh | 16422 | 3120 | 37 | 14 | 561 | 258 | 558 | 535 | 122 | 74 | 21671 |
| Curated papers | 246 | 56 | 11 | 2 | 1 | 9 | 2 | 6 | 1 | 1 | 335 |
aTO: traits—the number of unique phenotypic traits defined by TO terms that have been used to annotate QTLs.
bTO: trait categories—the nine categories of traits named in the TO; this higher order node in the TO serves to cluster related traits.
cPO: structure—the number of anatomy terms used to describe QTLs (total number of unique terms = 48).
dPO: growth-stage—the number of growth-stage terms used to describe QTLs (total number of unique terms = 20).
eMap Sets—the number of unique mapping population marker datasets used in QTL studies.
fParental germplasm—the number of different strains or accessions used as parents in QTL mapping studies.
gCo-localized markers—markers that map within QTL intervals; >37 000 markers have been curated and used to anchor QTLs to the sequence map of rice.
hNeighboring markers—markers flanking QTL intervals; >21 000 neighboring markers have been curated and are used to construct comparative maps.
Summary of QTL and associated features in the Gramene database for 10 cereal species
| Updated based on build 28. | Rice | Maize | Wheat | Tetraploid wheat | Oat | Barley | Sorghum | Pearl millet | Foxtail millet | Wild rice | Total |
|---|---|---|---|---|---|---|---|---|---|---|---|
| QTLs | 8646 | 1747 | 23 | 8 | 375 | 299 | 136 | 284 | 65 | 41 | 11624 |
| TO: traitsa | 237 | 77 | 10 | 3 | 7 | 30 | 19 | 27 | 2 | 10 | 332 |
| TO: trait categoriesb | 9 | 8 | 3 | 2 | 5 | 7 | 5 | 6 | 1 | 5 | 9 |
| PO: structurec | 38 | 19 | 5 | 2 | 6 | 8 | 7 | 15 | 2 | 5 | 48 |
| PO: growth-staged | 19 | 9 | 6 | 2 | 5 | 5 | 7 | 7 | 2 | 5 | 20 |
| Map setse | 89 | 8 | 9 | 2 | 1 | 8 | 2 | 5 | 1 | 1 | 126 |
| Parental germplasmf | 91 | 4 | 4 | 2 | 2 | 7 | 3 | 12 | 2 | 2 | 108 |
| Co-localized markersg | 30950 | 3615 | 73 | 14 | 888 | 335 | 334 | 1031 | 87 | 42 | 37369 |
| Neighboring markersh | 16422 | 3120 | 37 | 14 | 561 | 258 | 558 | 535 | 122 | 74 | 21671 |
| Curated papers | 246 | 56 | 11 | 2 | 1 | 9 | 2 | 6 | 1 | 1 | 335 |
| Updated based on build 28. | Rice | Maize | Wheat | Tetraploid wheat | Oat | Barley | Sorghum | Pearl millet | Foxtail millet | Wild rice | Total |
|---|---|---|---|---|---|---|---|---|---|---|---|
| QTLs | 8646 | 1747 | 23 | 8 | 375 | 299 | 136 | 284 | 65 | 41 | 11624 |
| TO: traitsa | 237 | 77 | 10 | 3 | 7 | 30 | 19 | 27 | 2 | 10 | 332 |
| TO: trait categoriesb | 9 | 8 | 3 | 2 | 5 | 7 | 5 | 6 | 1 | 5 | 9 |
| PO: structurec | 38 | 19 | 5 | 2 | 6 | 8 | 7 | 15 | 2 | 5 | 48 |
| PO: growth-staged | 19 | 9 | 6 | 2 | 5 | 5 | 7 | 7 | 2 | 5 | 20 |
| Map setse | 89 | 8 | 9 | 2 | 1 | 8 | 2 | 5 | 1 | 1 | 126 |
| Parental germplasmf | 91 | 4 | 4 | 2 | 2 | 7 | 3 | 12 | 2 | 2 | 108 |
| Co-localized markersg | 30950 | 3615 | 73 | 14 | 888 | 335 | 334 | 1031 | 87 | 42 | 37369 |
| Neighboring markersh | 16422 | 3120 | 37 | 14 | 561 | 258 | 558 | 535 | 122 | 74 | 21671 |
| Curated papers | 246 | 56 | 11 | 2 | 1 | 9 | 2 | 6 | 1 | 1 | 335 |
aTO: traits—the number of unique phenotypic traits defined by TO terms that have been used to annotate QTLs.
bTO: trait categories—the nine categories of traits named in the TO; this higher order node in the TO serves to cluster related traits.
cPO: structure—the number of anatomy terms used to describe QTLs (total number of unique terms = 48).
dPO: growth-stage—the number of growth-stage terms used to describe QTLs (total number of unique terms = 20).
eMap Sets—the number of unique mapping population marker datasets used in QTL studies.
fParental germplasm—the number of different strains or accessions used as parents in QTL mapping studies.
gCo-localized markers—markers that map within QTL intervals; >37 000 markers have been curated and used to anchor QTLs to the sequence map of rice.
hNeighboring markers—markers flanking QTL intervals; >21 000 neighboring markers have been curated and are used to construct comparative maps.
Distribution of rice QTLs
| Chr. | Chr. length (Mb) | No. of QTLs | QTLs with genome positiona | QTLs associated with Trait category term, Vigorb | QTLs associated with plant height |
|---|---|---|---|---|---|
| 1 | 43.6 | 1274 | 1026 | 255 | 157 |
| 2 | 35.9 | 843 | 590 | 96 | 44 |
| 3 | 36.3 | 1069 | 755 | 181 | 102 |
| 4 | 35.2 | 769 | 500 | 113 | 60 |
| 5 | 29.9 | 667 | 512 | 99 | 53 |
| 6 | 31.2 | 826 | 632 | 84 | 30 |
| 7 | 29.7 | 653 | 448 | 82 | 28 |
| 8 | 28.3 | 646 | 453 | 78 | 31 |
| 9 | 23.0 | 519 | 392 | 78 | 46 |
| 10 | 22.9 | 397 | 312 | 48 | 31 |
| 11 | 28.5 | 546 | 422 | 92 | 40 |
| 12 | 27.5 | 437 | 251 | 49 | 16 |
| Total | 372.0 | 8646 | 6293 | 1255 | 638 |
| Chr. | Chr. length (Mb) | No. of QTLs | QTLs with genome positiona | QTLs associated with Trait category term, Vigorb | QTLs associated with plant height |
|---|---|---|---|---|---|
| 1 | 43.6 | 1274 | 1026 | 255 | 157 |
| 2 | 35.9 | 843 | 590 | 96 | 44 |
| 3 | 36.3 | 1069 | 755 | 181 | 102 |
| 4 | 35.2 | 769 | 500 | 113 | 60 |
| 5 | 29.9 | 667 | 512 | 99 | 53 |
| 6 | 31.2 | 826 | 632 | 84 | 30 |
| 7 | 29.7 | 653 | 448 | 82 | 28 |
| 8 | 28.3 | 646 | 453 | 78 | 31 |
| 9 | 23.0 | 519 | 392 | 78 | 46 |
| 10 | 22.9 | 397 | 312 | 48 | 31 |
| 11 | 28.5 | 546 | 422 | 92 | 40 |
| 12 | 27.5 | 437 | 251 | 49 | 16 |
| Total | 372.0 | 8646 | 6293 | 1255 | 638 |
aThe number of QTLs that have positions on the sequenced rice genome is less than the total number of QTLs curated due to lack of requisite information in some publications (i.e. for AFLP, RAPD, etc.).
bTO trait category term ‘Vigor’ carries annotations for traits related to rate of seedling emergence, biomass accumulation, etc.
Distribution of rice QTLs
| Chr. | Chr. length (Mb) | No. of QTLs | QTLs with genome positiona | QTLs associated with Trait category term, Vigorb | QTLs associated with plant height |
|---|---|---|---|---|---|
| 1 | 43.6 | 1274 | 1026 | 255 | 157 |
| 2 | 35.9 | 843 | 590 | 96 | 44 |
| 3 | 36.3 | 1069 | 755 | 181 | 102 |
| 4 | 35.2 | 769 | 500 | 113 | 60 |
| 5 | 29.9 | 667 | 512 | 99 | 53 |
| 6 | 31.2 | 826 | 632 | 84 | 30 |
| 7 | 29.7 | 653 | 448 | 82 | 28 |
| 8 | 28.3 | 646 | 453 | 78 | 31 |
| 9 | 23.0 | 519 | 392 | 78 | 46 |
| 10 | 22.9 | 397 | 312 | 48 | 31 |
| 11 | 28.5 | 546 | 422 | 92 | 40 |
| 12 | 27.5 | 437 | 251 | 49 | 16 |
| Total | 372.0 | 8646 | 6293 | 1255 | 638 |
| Chr. | Chr. length (Mb) | No. of QTLs | QTLs with genome positiona | QTLs associated with Trait category term, Vigorb | QTLs associated with plant height |
|---|---|---|---|---|---|
| 1 | 43.6 | 1274 | 1026 | 255 | 157 |
| 2 | 35.9 | 843 | 590 | 96 | 44 |
| 3 | 36.3 | 1069 | 755 | 181 | 102 |
| 4 | 35.2 | 769 | 500 | 113 | 60 |
| 5 | 29.9 | 667 | 512 | 99 | 53 |
| 6 | 31.2 | 826 | 632 | 84 | 30 |
| 7 | 29.7 | 653 | 448 | 82 | 28 |
| 8 | 28.3 | 646 | 453 | 78 | 31 |
| 9 | 23.0 | 519 | 392 | 78 | 46 |
| 10 | 22.9 | 397 | 312 | 48 | 31 |
| 11 | 28.5 | 546 | 422 | 92 | 40 |
| 12 | 27.5 | 437 | 251 | 49 | 16 |
| Total | 372.0 | 8646 | 6293 | 1255 | 638 |
aThe number of QTLs that have positions on the sequenced rice genome is less than the total number of QTLs curated due to lack of requisite information in some publications (i.e. for AFLP, RAPD, etc.).
bTO trait category term ‘Vigor’ carries annotations for traits related to rate of seedling emergence, biomass accumulation, etc.
QTL map sets, markers and parental crosses
Gramene's QTL database now contains 126 QTL Map Sets for 10 different species with 11 624 QTLs, 40 553 markers, 37 369 QTL co-localized markers and 21 671 QTL neighboring markers. The QTL Map Sets are valuable online resources that are extensively used by researchers seeking information about the allelic composition of diverse germplasm resources and mapping populations. Table 3 summarizes information about selected Map Sets (of the 126) from five different species and highlights specific features associated with each.
Sample of information associated with seven QTL map sets from five species in Gramene
| Map set name | Species | QTL | No. of markers on map | Co-localized markers | Neighboring markers | Parental germplasm | No. of associated QTL studies |
|---|---|---|---|---|---|---|---|
| 1. Cornell IR64/Azu DH QTL 2001 | Rice | 275 | 588 | 287 | 203 | IR64, Azucena | 9 |
| 2. IRRI IR64/Azu DH QTL 2003 | Rice | 1372 | 281 | 2529 | 2636 | IR64, Azucena | 23 |
| 3. JRGP Nip/Kas F2 QTL 2000 | Rice | 351 | 3263 | 12732 | 1185 | Nipponbare, Kasalath | 33 |
| 4. UWM B73/Mo17 RFLP SSR QTL 1996 | Maize | 66 | 186 | 125 | 120 | B73, Mo17 | 3 |
| 5. Synthetic/Opata RI RFLP/SSR QTL 1995 | Wheat | 1 | 943 | 1 | 1 | Synthetica, Opata | 4 |
| 6. Steptoe/Morex DH RFLP QTL 2006 | Barley | 33 | 312 | 1 | 66 | Steptoe, Morex | 1 |
| 7. Cornell Kan/Ogl QTL 1995 | Oat | 375 | 249 | 888 | 528 | Kanota, Ogle | 1 |
| Map set name | Species | QTL | No. of markers on map | Co-localized markers | Neighboring markers | Parental germplasm | No. of associated QTL studies |
|---|---|---|---|---|---|---|---|
| 1. Cornell IR64/Azu DH QTL 2001 | Rice | 275 | 588 | 287 | 203 | IR64, Azucena | 9 |
| 2. IRRI IR64/Azu DH QTL 2003 | Rice | 1372 | 281 | 2529 | 2636 | IR64, Azucena | 23 |
| 3. JRGP Nip/Kas F2 QTL 2000 | Rice | 351 | 3263 | 12732 | 1185 | Nipponbare, Kasalath | 33 |
| 4. UWM B73/Mo17 RFLP SSR QTL 1996 | Maize | 66 | 186 | 125 | 120 | B73, Mo17 | 3 |
| 5. Synthetic/Opata RI RFLP/SSR QTL 1995 | Wheat | 1 | 943 | 1 | 1 | Synthetica, Opata | 4 |
| 6. Steptoe/Morex DH RFLP QTL 2006 | Barley | 33 | 312 | 1 | 66 | Steptoe, Morex | 1 |
| 7. Cornell Kan/Ogl QTL 1995 | Oat | 375 | 249 | 888 | 528 | Kanota, Ogle | 1 |
a‘Synthetic’ means that the female parent is Altar84/A3. squarrose (219) IGM86.940.
Sample of information associated with seven QTL map sets from five species in Gramene
| Map set name | Species | QTL | No. of markers on map | Co-localized markers | Neighboring markers | Parental germplasm | No. of associated QTL studies |
|---|---|---|---|---|---|---|---|
| 1. Cornell IR64/Azu DH QTL 2001 | Rice | 275 | 588 | 287 | 203 | IR64, Azucena | 9 |
| 2. IRRI IR64/Azu DH QTL 2003 | Rice | 1372 | 281 | 2529 | 2636 | IR64, Azucena | 23 |
| 3. JRGP Nip/Kas F2 QTL 2000 | Rice | 351 | 3263 | 12732 | 1185 | Nipponbare, Kasalath | 33 |
| 4. UWM B73/Mo17 RFLP SSR QTL 1996 | Maize | 66 | 186 | 125 | 120 | B73, Mo17 | 3 |
| 5. Synthetic/Opata RI RFLP/SSR QTL 1995 | Wheat | 1 | 943 | 1 | 1 | Synthetica, Opata | 4 |
| 6. Steptoe/Morex DH RFLP QTL 2006 | Barley | 33 | 312 | 1 | 66 | Steptoe, Morex | 1 |
| 7. Cornell Kan/Ogl QTL 1995 | Oat | 375 | 249 | 888 | 528 | Kanota, Ogle | 1 |
| Map set name | Species | QTL | No. of markers on map | Co-localized markers | Neighboring markers | Parental germplasm | No. of associated QTL studies |
|---|---|---|---|---|---|---|---|
| 1. Cornell IR64/Azu DH QTL 2001 | Rice | 275 | 588 | 287 | 203 | IR64, Azucena | 9 |
| 2. IRRI IR64/Azu DH QTL 2003 | Rice | 1372 | 281 | 2529 | 2636 | IR64, Azucena | 23 |
| 3. JRGP Nip/Kas F2 QTL 2000 | Rice | 351 | 3263 | 12732 | 1185 | Nipponbare, Kasalath | 33 |
| 4. UWM B73/Mo17 RFLP SSR QTL 1996 | Maize | 66 | 186 | 125 | 120 | B73, Mo17 | 3 |
| 5. Synthetic/Opata RI RFLP/SSR QTL 1995 | Wheat | 1 | 943 | 1 | 1 | Synthetica, Opata | 4 |
| 6. Steptoe/Morex DH RFLP QTL 2006 | Barley | 33 | 312 | 1 | 66 | Steptoe, Morex | 1 |
| 7. Cornell Kan/Ogl QTL 1995 | Oat | 375 | 249 | 888 | 528 | Kanota, Ogle | 1 |
a‘Synthetic’ means that the female parent is Altar84/A3. squarrose (219) IGM86.940.
QTLs are displayed in two ways in Gramene; using the Generic Model Organism Database (GMOD) CMap application, a comparative map-viewing tool that can display multiple maps simultaneously (http://gmod.org/wiki/CMap), and using the Ensembl-based Genome Browser (2), which presents QTL locations in the context of the reference genome and related annotations.
Annotation of QTL using ontologies
The problem of semantic heterogeneity in published descriptions of phenotypes has been a long-standing challenge to the scientific community. Free-text descriptions present a major hurdle in data management, and within a genome database, descriptive information must be normalized or standardized so that it is machine-readable before it can be associated with other features in the genome. This is in stark contrast to the standardized and highly automated representations of genomic and protein sequence data. Because of the large scale of data that must be annotated and the complexity of the associations among data types within the database, Gramene curators and developers have participated in or pioneered the development of several key ontologies (10–12,16).
Ontologies provide a network of controlled vocabularies, linked by relationships that reflect the biological realities they describe, that are used to facilitate machine-based queries that can rapidly extract information from a database. Terms from the TO, PO, GSO and EO were used to record the essential phenotypic information from each study. The TO currently consists of 973 terms or concepts that are used to describe plant traits or phenotypic assays and in the current version of the database 344 TO terms have been used to annotate QTL (Table 1). The PO has two aspects, one pertains to plant anatomy and the other to plant growth stages. There are over 853 terms in the PO and 275 terms in the GSO, but most QTLs have been assayed only in a small number of all possible tissues, organs and growth stages. Currently, 49 unique anatomical terms and about 15 distinct stages of the rice life cycle are associated with the curated QTLs (Table 1). The EO has over 496 terms descriptive of the major environments in which plants are grown, indicating temperature, water and day-length regimes, nutrient status, pest and pathogen exposure, and whether experiments were conducted under greenhouse, growth chamber or field conditions, etc. However, most historical QTL studies provide little information about the specific environmental parameters that defined the growth conditions of plants, and as a result, only about 27 EO terms have been used to annotate the QTLs in Gramene, many of which are related to specific biotic or abiotic stresses imposed as treatments.
Annotation of QTLs with terms from different ontologies enhances the querying potential of the database (please see tutorial pdf for step by step use of the TO and QTL database within Gramene at http://www.gramene.org/tutorials/QTL_tutorial.pdf). For example, TO annotation supports queries such as, ‘Show all the QTLs associated with flowering time or seedling vigor in rice’ or ‘Show all the flowering time QTLs from rice, barley and maize’. Furthermore, because every QTL is defined as a location on the genome, users can ask, ‘Show all the QTL associated with flowering time between marker X and marker Y on chromosome 6 of rice’. Annotation with PO terms enables users to ask, ‘Show all the QTLs associated with the “spikelet meristem” or with “embryo development” ’(10). The EO makes it possible to query QTLs associated with a plant's response to a specific environmental condition, such as bacterial blight or day length.
Display and navigation features
Entry
The main entry point to the QTL module of Gramene is through the ‘QTL’ link on the Gramene Navigation bar. A user can search QTLs by trait name, trait symbol, trait category, linkage group, published QTL symbol, Gramene QTL accession number or by associations such as QTL-associated genes and markers or PO terms. The QTL search interface transmits a user's specifications for a QTL to the database and renders the responses as individual web pages containing a downloadable subset of QTL records or detailed information page for a specific QTL.
Links to other modules
The QTL database has links to other modules such as the marker, maps, genome browser, ontology, literature, gene and genetic diversity databases. In addition, links are provided to other databases whose QTL data are hosted in Gramene, such as MaizeGDB and GrainGenes.
Miscellaneous
The entire database currently has two cycles of data release per year, and the QTL release notes are updated and the current statistics are available at ftp://ftp.gramene.org/pub/gramene/CURRENT_RELEASE/data/statistics/qtl_statistics/
An extensive PDF tutorial helps new users navigate through the QTL database (http://www.gramene.org/tutorials/QTL_tutorial.pdf). The feedback button on the QTL page allows users to communicate with the Gramene curators and developers. Feedback queries are usually responded to within 48 h.
Generalized model for using the QTL module for gene discovery
Forward genetics
In the following example, we will demonstrate how to integrate data and information from Gramene to facilitate the identification of genes or regulatory sequences underlying QTLs based on a forward genetics approach.
Plant height is a complex trait that reflects a plant's ability to respond to both internal developmental signals and exogenous stimulants in the environment. It has been extensively studied in cereals and is widely used as an indicator of plant vigor in the field and as a criterion for selection in plant breeding programs. In our example, a rice geneticist has identified a plant height QTL, spanning a 39.2 cM interval on chromosome 9, flanked by two RFLP markers (RZ206 and RZ12) on the published IR64/Azucena QTL map (19,20) The QTL has been curated as ‘CQAX26’ and can be searched using this ID in the database. We want to use the Gramene database to digitally fine map the QTL and move from a QTL interval on a genetic map, to its position on a physical or a sequence map, to a list of candidate genes.
By querying on ‘CQAX26’, and following the links, a user finds information about the QTL, including ‘Map Positions’, ‘Associated Markers’, ‘Associated Genes’, ‘Associated Ontologies’ and ‘Database Cross-Reference’. Clicking on ‘Map Positions’ provides information about the location of the QTL on the Sequence map, the physical size of the region encompassed by the QTL (7.62 Mb) and links to genetic (QTL) maps. In the QTL map type, we can open the link to the ‘CMap Preview’ to get a view of all the QTLs associated with chromosome 9 on the IR64/Azucena mapping population and allows a user to see the position of the markers flanking the plant height QTL (RZ206, RZ422, RZ228 and RZ12) aligned to the sequence map of rice (Supplementary Figure 1). In the Sequence map type, one can click on ‘View in Genome Browser’ to identify numerous molecular markers in the region, including over 100 SSR and over a dozen RFLP (Supplementary Figure 2a). These may be useful if the researcher wants to experimentally fine map the QTL. For example, one can click on a marker of interest and open up a page from the Gramene Markers database to obtain information on primer pairs, PCR conditions or other properties of marker assays (Supplementary Figure 2b). Using the available markers and sequence information, a researcher can undertake a wet-lab experiment to look for informative recombinants in a large, fine mapping population.
As the fine mapping proceeds, Gramene's Genome Browser can be used to provide a high-resolution view of a specific portion of rice chromosome 9 (Supplementary Figure 3), so that the precise position of flanking markers can be clarified in relation to a set of candidate genes. From the detailed view of the region, she/he can identify gene models that comprise the first pass list of candidate genes.
Based on GO ontology annotations and the BLAST search tool available in Gramene, the list of gene sequences can be used to look for homologs in other model organisms, such as Arabidopsis or maize. In many cases, the candidate genes can be associated with biochemical pathways (http://www.gramene.org/pathway/) and/or with putative gene functions based on GO annotations (http://www.gramene.org/plant_ontology/index.html). Gramene also provides information about natural and induced mutations [i.e. from functional genomics populations that have flanking sequence tags (FSTs) marking the mutations] that are associated with genes of known function and/or measurable phenotypic variation (Supplementary Figure 4). Functional mutations with known map positions can be aligned to determine which, if any, fall within the target QTL region.
A close look at the gene models that fall within the plant height QTL on chromosome 9 shows that four of the predicted genes in this region are rational candidates for the gene(s) underlying the plant height QTL, CQX26. Among them, three (LOC_Os09g24800, LOC_Os09g24810 and LOC_Os09g24840) are homologs of Arabidopsis genes that have been annotated with the GO terms, ‘giberrellin-related’ or ‘brassinosteroid-related’ (Supplementary Figure 5), both of these terms are known to be associated with pathways related to plant growth and development. In addition, the gene LOC_Os09g25150 is part of the brassinosteroids pathway as annotated in RiceCyc, the pathways database within Gramene (http://pathway.gramene.org/RICE/server.html?). Another search within Gramene identified 7 T-DNA insertion lines (CL522872.1, CL523877.1, CL523487.1, etc.) (21) with FSTs that mapped within the small, fine mapped QTL region (14 704 764–15 064 206 bp).
The combination of positional information, mutants, markers, germplasm and phenotypes offers database users a variety of ways to approach the problem of identifying a gene(s) underlying a QTL. Mutant lines are likely to be of interest for the complementation experiments and functional analyses that are ultimately required to validate phenotype-gene hypotheses. The example outlined here clearly demonstrates how QTL data, together with other types of data and tools from Gramene, can be used to facilitate the job of narrowing down a QTL region, selecting and validating candidate genes that condition complex phenotypes (Figure 2). If no candidate genes are identified, the database will serve primarily as a resource for identifying sequence polymorphisms and for clarifying positional information so that fine mapping and positional cloning can proceed. Once the gene(s) or sequence variant(s) that is causally responsible for a quantitatively inherited phenotype has been conclusively demonstrated, the database will also serve as a repository for that information so that it can be leveraged by the next generation of users in their efforts to associate genotypic and phenotypic variation.
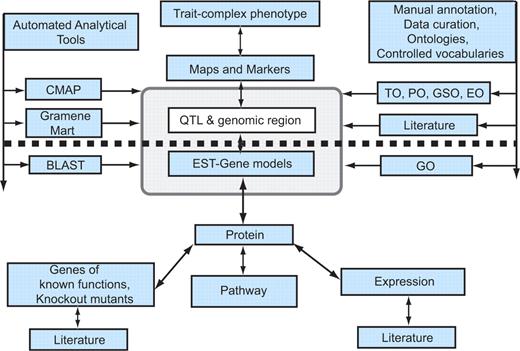
Generalized model for candidate gene discovery. The diagram shows a simplified view of the information modules in the Gramene database. The figure has been designed to show the path of forward genetic dissection of complex traits above the dotted line and reverse genetic investigation below the heavy dotted line. In reality, as indicated by the arrows, the database can be accessed from any point of entry and users can navigate to the required information. Gramene provides automated analytical tools shown to the left of the pale grey box, and manual curation using ontologies and controlled vocabularies shown to the right of the dotted box, to enhance the data mining potential of the database. The association between the QTL and EST modules contained within the pale grey box highlights the fact that a user can systematically zero in on information about the relationship between a complex phenotype and a candidate gene/s of interest. As depicted here, a trait or complex phenotype is associated through maps and markers to a genomic region identified as a QTL and to ESTs and gene models previously annotated to the genome region; the QTL database thus performs the crucial function of connecting molecular or genotypic information with complex phenotypes, and more generally connecting the field of molecular biology with that of quantitative genetics and plant breeding.
Reverse genetics
The next example illustrates database navigation from a reverse genetics perspective. In this example, we will focus on iron-uptake, a well-characterized metabolic pathway, in rice.
Iron is an essential element involved in almost all of the life-supporting physiological and cellular processes of plants. It is required for respiration, photosynthesis, DNA synthesis, N2 fixation and hormone production [for review, see Guerinot (22)]. Plants utilize two main, tightly regulated mechanisms of Fe-uptake: reduction and chelation. Several genes of these pathways including some transcription factors have been cloned and characterized (23,24).
Integration of information derived from biochemical and molecular studies into applied breeding programs presents several challenges. Mechanistic information is not easily translated into new varieties that succeed in the marketplace. This raises the question: can we transform what we learn about molecular mechanisms underlying critical biochemical pathways into predictive information about phenotypes that can be measured at the whole-plant level under field conditions? Is there a systematic approach to data mining that will allow us to extract clues about how to manipulate well-characterized biochemical pathways and gene networks to achieve a set of desired phenotypic consequences in the context of agricultural production?
Iron deficiency chlorosis (IDC) or loss of green pigment is the condition that affects plants unable to acquire and utilize iron. These plants develop foliar chlorosis of young leaves leading to yield loss, particularly in alkaline soils where the high pH reduces the solubility of iron. Thus, IDC in alkaline soils is caused not by a lack of iron, but rather by lack of iron uptake (25). In contrast, when rice is grown in flooded, anaerobic conditions or in acidic, paddy soils, excess accumulation of iron in plant roots can lead to iron toxicity. This causes severe damage to plants as the free iron catalyzes the formation of reactive oxygen species (26,27). In order to increase productivity in both anaerobic and aerobic soils, it is therefore necessary to select plants that respond appropriately to excess or deficiency of iron, and it is helpful to understand the iron-uptake pathway and its regulation to accomplish this goal.
Curated information about the genetics and biochemistry of the Fe-uptake pathway appears in several different sub-databases within Gramene, namely the Gene, Protein, Pathway and QTL databases. While several ‘iron toxicity’ QTLs have been published, there are no reports of QTLs for IDC in rice, despite the widespread occurrence of the problem (28,29).
We undertook a search using reverse genetics to try to identify a relationship between genes known to be important in the Fe-uptake pathways based on molecular and biochemical information, and traits or phenotypes associated with QTLs that were likely to be associated with the symptoms of IDC using the Gramene database. Our underlying hypothesis was that IDC may have been described in rice as a whole-plant phenotype in such a way that its connection to the Fe-uptake pathway might not be obvious (Figure 3).
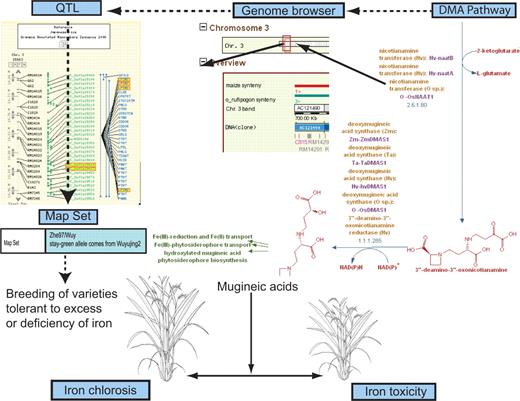
Iron-uptake pathway in rice: cloned gene to complex traits, via pathway, genes and QTLs. Leaf senescence QTLs aligned to CMap view of rice chromosome 3 shown in the top left corner; Genome Browser view of QTL region of rice chromosome 3 with associated gene models shown in the center; DMA metabolic pathway shown in the top right; rice plants expressing iron chlorosis tolerant phenotype and iron toxicity phenotype shown at the bottom. The Mugenic acids (MAs) or phytosiderophores are end products of the DMA metabolic pathway. Biosynthesis of MAs is closely linked to iron deficiency and is highly regulated to avoid toxicity owing to excess iron uptake. The figure represents parts of the curated DMA pathway; the genomic positions of the cloned genes from this pathway are used to trace suitable QTL candidates in that region. The populations from which these QTLs were analyzed can then be used in breeding programs aimed at developing rice varieties that are tolerant to soil environments characterized by deficiencies or excesses of iron.
To initiate our search, we looked at one of the main physiological processes related to iron chlorosis (photosynthesis and, in particular, loss of green pigmentation in the young leaves) and searched for QTLs with related phenotypic expression. The phytosiderophore biosynthesis pathway in rice referred to as the deoxymugineic acid (DMA) pathway has two critical enzymatic steps catalyzed by nicotianamine synthase (OsNAS1 = LOC_OsO3g19427; OsNAS2 = LOC_OsO3g19420; and OsNAS3 =LOC_OsO7g48980) and nicotianamine transferase (OsNAAT1 = LOC_OsO3g13390). We obtained gene sequences for these enzymes by searching the NCBI database and BLATed these gene sequences against the Nipponbare genome to determine their genomic positions. Using the physical position of the genes, we then searched the QTL database in Gramene to identify all QTLs that had intervals overlapping the gene models.
We obtain a list of all QTLs in the database by entering the QTL module and searching on the wild-card key (*). We can then filter by linkage group followed by a second filter on species to obtain only the QTLs on rice chromosome 3. This list can be downloaded and sorted by position to identify those of interest. Once this subset of QTLs has been identified, it can be searched by trait name to identify those related to iron chlorosis and the ‘co-localized’ and ‘neighboring’ markers listed in the QTL page can be used to align the genetic map (cM) with the sequence map (bp) (as illustrated in [Supplementary Figure 1]). More formally, we searched for an intersection between GO annotations assigned to the NAS and NAAT genes, and PO and TO annotations assigned to the QTLs, in an effort to identify QTLs that might rationally be associated with IDC. The most salient were QTLs associated with ‘leaf senescence’ that mapped to the OsNAS gene region on chromosome 3. The rationale behind this thinking is that (i) photosynthesis is the main physiological process associated with the DMA pathway, (ii) deficient Fe-uptake, mediated by the NAS and NAAT genes, results in impaired photosynthesis and (iii) impaired photosynthesis leads to leaf chlorosis and ultimately to leaf senescence. In support of this logic, recent studies have indicated that increased senescence is associated with gene expression induced by iron deficiency (30). Rice seedlings, grown in the absence of iron, induced genes that were classically associated with leaf senescence (31).
This example demonstrates how the QTL database can help integrate information in a way that allows researchers to explore relationships between diverse data domains, phenotypic descriptors, germplasm accessions, genes, proteins and pathways (Figure 3). It highlighted a logical, but digitally cryptic connection between the genetic mechanisms of IDC and those of leaf senescence. It provided the basis for asking whether we can use variation in leaf senescence observed under conditions of Fe-sufficiency and -deficiency to identify germplasm or populations that may be useful as donors of IDC resistance in a plant breeding program. In this way, the QTL module may help the research community begin to bridge the gap between our understanding of genetic mechanisms and our ability to identify germplasm resources that carry valuable alleles for crop improvement.
Discussion
Utility of Gramene's QTL database in gene discovery
The original concept of a QTL was first proposed by Sax (32), long before the advent of DNA marker technology. Today, one of the great challenges facing geneticists is to identify the genes or regulatory sequences underlying QTL and to understand their molecular functions and how they interact in biochemical and regulatory pathways to determine a phenotype (5). In developing the QTL module in Gramene, we confronted several of the major impediments facing the genetics community in achieving this goal: (i) information about QTL was deeply buried in hundreds of diverse publications or researchers’ files, making it very difficult for the research community to share information; (ii) different names, terms and concepts were used to define or describe anatomical parts, developmental stages and traits of interest across species and research groups; (iii) QTLs were mapped onto different genetic maps using different markers and mapping populations making it difficult to compare QTL locations across populations, maps and species; and (iv) the QTL intervals often comprise large genomic regions, typically covering 10–20 cM and may include up to ∼2000 genes (6), making it virtually impossible to identify the gene(s) underlying a QTL based on individual studies. Thus, fine mapping has been widely used to pinpoint QTL more accurately, i.e. to a region of <1cM containing <25 genes, but this strategy requires the investment of large amounts of time and resources and fails to leverage historical data effectively.
To address the challenges outlined above, Gramene curators developed a QTL resource that enhances researcher's abilities to more efficiently utilize previously published data to identify the genes underlying QTL. First, they collected publications on QTL in rice and related cereal species and developed a central literature repository. Next, QTL information was extracted manually from the published sources and was annotated using controlled vocabularies and free text. Using controlled vocabularies, agronomic traits and QTL associated with anatomy terms or developmental stages were carefully described and defined. This has made it possible to compare phenotypes associated with QTL among studies and across species.
For QTL, using flanking markers as mediators, QTLs originally reported on individual genetic maps have been systematically aligned to the rice sequence map, where they can now be searched as standard genomic features. With the rice genome as common platform, researchers are now able to determine whether a QTL of interest is co-localized with other QTLs associated with the same or similar traits detected in independent QTL experiments. The database also facilitates combining data from multiple datasets to improve the resolution of a QTL position. Researchers can more reliably identify a list of candidate genes that co-localize with the target QTL and they can subsequently select a short list of candidate genes using GO terms as functional delimitors. Eventually, expression and metabolic data from large, high-throughput experiments, such as microarray or protein data, can be integrated with QTL results. In addition, parental germplasm and progeny lines used in bi-parental QTL studies (double haploid lines, recombinant lines, near-isogenic lines), functional genomics populations with FSTs marking mutations of interest, and association mapping panels can also be jointly employed for further fine mapping and gene function validation. Although researchers rarely cite the databases used to facilitate gene discovery efforts, Gramene is widely used to support such activities (33,34).
Applications of the QTL database in plant breeding and selection
The QTL and trait-associated markers are crucially important for applications in plant breeding and selection. Two types of QTL-associated markers are curated in Gramene, co-localized QTL markers and neighboring QTL markers. In addition to this information, researchers can use Gramene to readily identify additional markers from corresponding regions on other maps and they can then align the regions of interest to each other and to the rice genome using CMap. From the list of available markers in the target region, researchers can then select a set of useful markers for use in fine mapping or to directly facilitate trait selection in a marker-assisted breeding program.
Gramene also provides information about the germplasm and genetic materials used in QTL studies. This type of information represents a useful resource for plant breeders who seek to identify parental germplasm and progeny lines containing favorable alleles or favorable recombination events between favorable and unfavorable alleles for particular agronomic traits. These lines may also serve as genetic materials for association mapping experiments. Lastly, using the sequence-based consensus map as a bridge, researchers are able to identify mutant lines, such as T-DNA insertion lines, where FST information indicates that a T-DNA element is likely to have altered the expression or function of a gene that maps within a QTL region of interest. Thus, Gramene offers a unique form of data integration, making it possible for researchers to readily find information about QTL-FST co-localization, and to bridge a major gap between genetic studies and breeding applications.
In this article, we have focused on the development and utilization of a QTL information resource that helps link molecular information with phenotypic information and traditional breeding applications. However, in addition to the manipulation of naturally occurring genetic variation through crossing and genetic recombination, the use of genetic engineering is of great interest to plant breeders. Gramene's QTL database provides valuable information to help identify candidate genes and superior alleles for use in both conventional crossing and selection schemes and for transgenic applications to plant improvement. The rational utilization of genetic diversity underlies all successful crop improvement strategies, and knowledge about the composition and the range of variation that exists both within and between species underlies plant breeders’ efforts to constructively manipulate genetic variation in an effort to achieve long-term productivity and sustainability of agricultural systems around the world.
Comparative power of the QTL database
Evolutionary studies of gene orthology and conserved synteny in the Poaceae (35–37) have supported the idea that grasses can be viewed as a single genetic system. The Gramene database is centered on this fundamental principle of phylogenomics. Recently, Gramene has expanded its coverage of the plant kingdom to include several key dicotyledonous species, making it an even more powerful comparative genomic resource for plants and broadening its extensive user base.
The falling cost and increasing efficiency of genotyping and resequencing technologies are fueling the generation of association mapping projects that generate datasets consisting of thousands or millions of SNPs evaluated on hundreds or thousands of individuals in many plant species. This deluge of diversity data provides the foundation for new approaches to comparative phenotypic, genotypic and expression analysis. As the Gramene database expands its schema to absorb this new data, the challenge of isolating genes underlying QTL becomes easier. Forward genetics approaches are facilitated by comparing phenotypic and genomic information from diverse species whose genomes can be aligned with rice, while reverse genetics approaches are enhanced by the growing reservoir of functional information about candidate genes from a wide range of species. CMap is used extensively for long-range comparative mapping, and the Genome Browser offers close-up views of conserved genes and regions. Phenotypic comparisons are greatly enhanced by the use of ontologies and improved data mining tools.
Future developments
Gramene's QTL database has evolved rapidly, from curating simple core QTL information to a model that involves annotation of multi-dimensional genetic diversity data using a variety of statistical approaches. Efforts are underway to collect and annotate raw phenotypic and genotypic data for several large-scale QTL and association mapping studies. Collection of raw data will enable researchers to apply publicly available software, such as TASSEL (Trait Analysis by aSSociation, Evolution and Linkage) (38,39) and R/qtl (http://www.rqtl.org/), to reanalyze existing datasets in an effort to capture new features through deep data mining, and to enhance the resolution of the analysis by integrating multiple QTL and association mapping datasets. Through this effort, geneticists will be able to more accurately identify peak markers for a particular QTL, identify breeding lines and genomic resources that contain favorable alleles for different traits, evaluate the breeding value of specific lines and quantify the percent of the phenotypic variation that is explained by particular genes or QTL in a given set of materials. Mining the data at this level will be greatly enhanced by Gramene's efforts to develop a large, multi-species repository of raw QTL data in a standardized format. In addition, we would like to partner with journals publishing QTL papers to develop a mechanism for reliable transfer/capture of published data into the Gramene database.
Additional efforts to enhance Gramene's analytical capacity to identify candidate genes underlying the QTLs and to map those genes to associated biochemical pathways are also underway. These efforts are based on developing algorithms and new statistical methods to facilitate gene discovery in a comparative context. These new approaches will enhance the utility of the information and strategies that are currently available.
Finally, we aim to enhance the querying capacity of the database and to provide batch download functionality. With Gramene Mart and other search interfaces to simplify the data mining and querying routines, users will soon be able to address increasingly complex queries regarding the inheritance, genetic mechanisms and evolution of quantitatively inherited traits.
Funding
National Science Foundation, Plant Genome Research Program (DBI 0703908 to L.S; subaward to S.McC.).
Conflict of interest: None declared.
Acknowledgements
We appreciate the numerous authors of QTL publications who have kindly shared information with us on QTL, markers and maps by way of personal communication. We are grateful to Carolyn Lawrence and Mary Schaeffer in MaizeGDB, Dave Matthews and Victoria Carollo in GrainGenes, Yukiko Yamazaki in Oryzabase and Richard Bruskiewich and Graham McLaren in IRIS, for their collaboration and sharing of data. We are grateful to Lois Swales for formatting.
References
Author notes
†Present address: Junjian Ni, Syngenta Biotechnology Inc., 3054 Cornwallis Road, Research Triangle Park, NC 27709, USA.
‡Present address: Anuradha Pujar, Boyce Thompson Institute, Cornell University, Ithaca, NY 14853-1801, USA.
¶Present address: Pankaj Jaiswal, Department of Botany and Plant Pathology, Oregon State University, Corvallis, OR 97331-2902, USA.

{kind=link}
{kind=link}
{kind=link}