





Abstract
Although research on influenza lasted for more than 100 years, it is still one of the most prominent diseases causing half a million human deaths every year. With the recent observation of new highly pathogenic H5N1 and H7N7 strains, and the appearance of the influenza pandemic caused by the H1N1 swine-like lineage, a collaborative effort to share observations on the evolution of this virus in both animals and humans has been established. The OpenFlu database (OpenFluDB) is a part of this collaborative effort. It contains genomic and protein sequences, as well as epidemiological data from more than 27 000 isolates. The isolate annotations include virus type, host, geographical location and experimentally tested antiviral resistance. Putative enhanced pathogenicity as well as human adaptation propensity are computed from protein sequences. Each virus isolate can be associated with the laboratories that collected, sequenced and submitted it. Several analysis tools including multiple sequence alignment, phylogenetic analysis and sequence similarity maps enable rapid and efficient mining. The contents of OpenFluDB are supplied by direct user submission, as well as by a daily automatic procedure importing data from public repositories. Additionally, a simple mechanism facilitates the export of OpenFluDB records to GenBank. This resource has been successfully used to rapidly and widely distribute the sequences collected during the recent human swine flu outbreak and also as an exchange platform during the vaccine selection procedure. Database URL: http://openflu.vital-it.ch.
Introduction
Influenza is a major disease caused by an RNA virus affecting a wide variety of hosts. Every year, infection by the traditional A/H1N1, A/H3N2 or B types of influenza cause ∼500 000 human deaths worldwide (1). Continuous genetic drift of its genome due to the error-prone RNA replication machinery (2) imposes a yearly re-evaluation of the vaccine composition (3). Occasionally, virus reassortants emerge as new strains which might cause dramatic pandemics such as the Spanish flu in 1918 (30–50 mio victims), the Asian flu in 1957 and the Hong Kong flu in 1969 (4). In the last decade, the highly pathogenic H5N1 and H7N7 avian influenza viruses have been sporadically transmitted from bird to human, which resulted in clinically severe and fatal infections (5,6). More recently, the triple-reassortant swine-like A/(H1N1) that is composed of RNA segments of viruses infecting swine, avian and human hosts has emerged in the human population (7,8). These observations result in a worldwide awakening on the possible raise of a new pandemic (6,9). The full support of the international scientific community is therefore urgently required to better understand the spread and evolution of the virus, and the determinants of its transmissibility and pathogenicity in humans. This in turn demands that scientists from different backgrounds of expertise have full access to comprehensive genetic, clinical and epidemiological data from both animal and human virus isolates in a timely manner.
Several efforts have been made to improve both comprehensiveness of influenza data records and information on the virus dissemination rate. The major existing influenza-specific resources include the Influenza virus resource (10) from NCBI, the Influenza Research Database from BioHealthBase (11), Influenza virus database (12) and Influenza Sequence and Epitope Database (13). They all operate at some level of openness and provide technical means for data exchange and analysis. The NCBI Influenza virus resource is a major influenza sequence repository that all the other listed references link to. It is a sequence-centric database that provides convenient standard sequence analysis tools (e.g. alignments, phylogenetic trees BLAST), biological annotation such as antiviral resistance, as well as basic epidemiological annotations. As influenza viruses are able to exchange genetic segments and switch from one host to another, it is essential to collect data from all types and subtypes on a wide variety of host species to track the origin of new viral strains. The OpenFlu database (OpenFluDB) was developed with a goal to collect viral sequences, as well as their detailed clinical and epidemiological metadata at the isolate level from all over the world, to annotate and to serve all these data back to the scientific community together with both sequence and epidemiological data mining tools. The database was launched in May 2008, and is free. More than a 1000 researchers of the influenza community or people interested in progress in the field have already used this resource.
As of January 2010, the database contains more than 27 000 isolates and 99 000 sequences. The data deposited within OpenFluDB can be pushed towards Genbank/DDBJ/EMBL depending on the laboratory and political body policy.
The OpenFluDB contents
OpenFluDB is isolate-centric, rather than sequence-centric and thus differs from the NCBI Influenza virus resource (10), the Influenza virus database (12) or the Influenza Sequence and Epitope Database (13). This choice facilitates the association between a comprehensive amount of clinical and epidemiological annotations with the viral sequences. In its present status, OpenFluDB contains data from A and B type viruses. Each virus isolate can be associated with the name of the institution providing the sample, the name of the laboratory that sequenced it and the name of the institution that submitted the data. General information about an isolate includes type, subtype, lineage, passage history, host, and collection date and place. Several clinical data including host age, sex or vaccination status and epidemiological information including in vivo-tested antiviral resistance can also be attributed to a virus strain. In addition, automatic annotations derived from sequence motif identification are used to computationally predict antiviral resistance, enhanced pathogenicity or putative human adaptation. This database stores both nucleotide sequences and automatically translated protein sequences.
Overview of the OpenFluDB user interface
The OpenFluDB user interface is composed of three parts. The first one, ‘Browse’, is designed to efficiently retrieve a set of isolates and related sequences that can be then submitted to several analysis tools like sequence similarity search and multiple sequence alignment (MSA), or mapped on geographical and sequence similarity maps (SSMs). Isolate records can be exported in Microsoft Excel format, and the nucleotide and protein sequences in FASTA format. The second part of the interface is the ‘Upload’ section, where users can deposit data either as a single isolate together with its sequences using a simple web form or a group of isolates by providing a properly formatted Microsoft Excel file together with the related sequences in a FASTA file. Subsequently, uploaded isolates can be easily exported to NCBI GenBank. The third part of the interface contains three different statistical views of the database content as histogram plots, geographical positions on a world map and SSMs produced by multidimensional scaling. Additionally, a ‘Help’ section collects a set of frequently asked questions and some screencasts illustrating several functionalities of the database.
Data upload
Users can populate OpenFluDB via two mechanisms: single isolate upload or batch upload. Uploading isolates one by one allows fine-grained control over the process, but is usually time-consuming. Upload by batch is more time efficient, but requires much user attention when preparing data. In addition, a daily automatic procedure imports isolates from GenBank. The uniqueness of one isolate is assessed by its combination of name and passage history. New sequences are annotated by automatic procedures.
Single isolate upload. This interface is meant to annotate one isolate at a time in a comprehensive way (Figure 1). It is a combination of several drop-down menus and text fields. Consistency of the data is improved by cross-checking of the different components of the isolate name with sample collection year and host annotation fields, as well as with a set of controlled vocabulary terms displayed as select menus. The geographical classification is adapted from GeoNames (14) and the taxonomical classification from NCBI taxonomy (15). Both classifications are displayed in form of hierarchical select menus to provide a convenient and consistent way of defining sample collection location and host species. Once an isolate is created, genetic sequences can be appended using either a web form for a single entry or by uploading multiple sequences from a FASTA file.

‘Single isolate upload’ interface. The web form is split into three parts. The top one contains fields to specify isolate name, isolate type and passage history. The syntax of the isolate name is verified against sample collection year and standard influenza nomenclature (except for laboratory-derived strains). The middle part contains sample data including host and sample collection date and geographical location. The bottom part contains additional data including sample provider laboratory, sequencer laboratory, in vivo tested antiviral resistance and user note.
Batch upload. Multiple isolates and sequences can be uploaded in batch using the combination of a Microsoft Excel file in a fixed format and a text file containing nucleotide sequences in FASTA format. The Excel template, meant to assist users to precisely describe isolates, can be downloaded from the ‘batch upload’ page. Each column header contains a short description of the required content and format. If the batch upload procedure fails because of inconsistency in annotation or improper sequences, a pair of Excel and FASTA files containing only the erroneous entries together with a comment on the errors is returned to the user for correction and resubmission.
Upon successful completion of single or batch upload procedure, a summary email is sent to the user listing all assigned OpenFluDB isolate identifiers (EPI_ISL_ID) and OpenFluDB sequence identifiers (EPIID). The newly uploaded data are immediately available to all registered users. Uploaded isolates can easily be exported to GenBank, where specific OpenFluDB annotations are kept in a structured comment.
Import. Besides user data uploads, daily automatic import procedures run to incorporate new influenza sequences and their annotation records from GenBank. Host, sample collection geographical location and sample collection year are extracted either from the respective annotation field or from the isolate name. Isolate and segment annotation consistency is checked. Imported GenBank entries that failed the automatic parsing procedure are not shown to users, but stored for subsequent manual curation.
New sequences from user upload or GenBank import are compared using BLAST (16) to a set of reference sequences to compute, and verify, user submitted values for virus type, subtype, lineage (for B type viruses) and segment name. Prediction of coding regions is performed on these nucleotide sequences; these are then translated into protein sequences that in turn are used to automatically detect antiviral resistance, high pathogenicity and human adaptation motifs. The criteria used to evaluate these features are described in (17–46) and summarized in Table 1.
List of mutations used to compute antiviral resistance and putative human adaptation
| Segment | Type (subtype) | Number of isolates | Position (H3 and N2 numbering) | Wild | Mutant | Antiviral resistance | High pathogenicity | Hum. adapt. | Ref. | |||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Neuraminidase inhibitors | Adam. | |||||||||||
| Ose. | Zan. | Per. | ||||||||||
| NA | A(N2), B | 12 | 119 | E | V | X | (38,40,42,43) | |||||
| NA | A(N2), B | 1 | 119 | E | A | X | X | (42,43) | ||||
| NA | A(N2), B | 3 | 119 | E | G | X | (42,43) | |||||
| NA | A(N2), B | 7 | 119 | E | D | X | X | (42,43) | ||||
| NA | A(N1) | 415 | 274 | H | Y | X | X | (34–38,41,43,44) | ||||
| NA | A(N1,N2) | 0 | 294 | N | S | X | (33) | |||||
| NA | A(N2), B | 2 | 292 | R | K | X | X | X | (39,42,43) | |||
| NA | B | 0 | 152 | R | K | X | X | X | (36–38,42,43) | |||
| NA | B | 3 | 198 | D | N/E | X | X | (38,43,44) | ||||
| NA | B | 1 | 222 | I | T | X | X | (44) | ||||
| NA | A(N2) | 6 | 222 | I | V | X | X | (44) | ||||
| M2 | A | 20 | 26 | L | F | X | (46) | |||||
| M2 | A | 130 | 27 | V | A | X | (45,46) | |||||
| M2 | A | 8 | 30 | A | T | X | (18,46) | |||||
| M2 | A | 1433 | 31 | S | N | X | (18,45,46) | |||||
| M2 | A | 0 | 34 | G | E | X | (46) | |||||
| HA | A | 2030 | HA cleavage site | ≥5 R or K | +++ | (24,25) | ||||||
| NP | A | 92 | 319 | N | K | ++ | (31) | |||||
| PB2 | A | 3181 | 627 | E | K | ++ | (29) | |||||
| PB2 | A | 177 | 701 | D | N | ++ | (29,30,31) | |||||
| NS1 | A | 97 | 92 | D | E | + (M) | + | (27,26) | ||||
| NS1 | A | 1240 | 80–84 | XXXXX | – | + (M) | + | (27) | ||||
| PB1-F2 | A | 1416 | 66 | N | S | ++ | (28) | |||||
| Segment | Type (subtype) | Number of isolates | Position (H3 and N2 numbering) | Wild | Mutant | Antiviral resistance | High pathogenicity | Hum. adapt. | Ref. | |||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Neuraminidase inhibitors | Adam. | |||||||||||
| Ose. | Zan. | Per. | ||||||||||
| NA | A(N2), B | 12 | 119 | E | V | X | (38,40,42,43) | |||||
| NA | A(N2), B | 1 | 119 | E | A | X | X | (42,43) | ||||
| NA | A(N2), B | 3 | 119 | E | G | X | (42,43) | |||||
| NA | A(N2), B | 7 | 119 | E | D | X | X | (42,43) | ||||
| NA | A(N1) | 415 | 274 | H | Y | X | X | (34–38,41,43,44) | ||||
| NA | A(N1,N2) | 0 | 294 | N | S | X | (33) | |||||
| NA | A(N2), B | 2 | 292 | R | K | X | X | X | (39,42,43) | |||
| NA | B | 0 | 152 | R | K | X | X | X | (36–38,42,43) | |||
| NA | B | 3 | 198 | D | N/E | X | X | (38,43,44) | ||||
| NA | B | 1 | 222 | I | T | X | X | (44) | ||||
| NA | A(N2) | 6 | 222 | I | V | X | X | (44) | ||||
| M2 | A | 20 | 26 | L | F | X | (46) | |||||
| M2 | A | 130 | 27 | V | A | X | (45,46) | |||||
| M2 | A | 8 | 30 | A | T | X | (18,46) | |||||
| M2 | A | 1433 | 31 | S | N | X | (18,45,46) | |||||
| M2 | A | 0 | 34 | G | E | X | (46) | |||||
| HA | A | 2030 | HA cleavage site | ≥5 R or K | +++ | (24,25) | ||||||
| NP | A | 92 | 319 | N | K | ++ | (31) | |||||
| PB2 | A | 3181 | 627 | E | K | ++ | (29) | |||||
| PB2 | A | 177 | 701 | D | N | ++ | (29,30,31) | |||||
| NS1 | A | 97 | 92 | D | E | + (M) | + | (27,26) | ||||
| NS1 | A | 1240 | 80–84 | XXXXX | – | + (M) | + | (27) | ||||
| PB1-F2 | A | 1416 | 66 | N | S | ++ | (28) | |||||
Ose., oseltamivir; Zan., zanamivir; Per., peramivir; Adam., adamantanes; Hum. adapt., human adaptation; Ref. bibliographic reference number; High pathogenicity could contribute, alone or in association with other mutations, to a higher pathogenicity in avians and/or mammals; +++, high level of confidence; ++, medium level of confidence; +, low level of confidence, or in correlation with other mutations; (M), mammals only.
List of mutations used to compute antiviral resistance and putative human adaptation
| Segment | Type (subtype) | Number of isolates | Position (H3 and N2 numbering) | Wild | Mutant | Antiviral resistance | High pathogenicity | Hum. adapt. | Ref. | |||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Neuraminidase inhibitors | Adam. | |||||||||||
| Ose. | Zan. | Per. | ||||||||||
| NA | A(N2), B | 12 | 119 | E | V | X | (38,40,42,43) | |||||
| NA | A(N2), B | 1 | 119 | E | A | X | X | (42,43) | ||||
| NA | A(N2), B | 3 | 119 | E | G | X | (42,43) | |||||
| NA | A(N2), B | 7 | 119 | E | D | X | X | (42,43) | ||||
| NA | A(N1) | 415 | 274 | H | Y | X | X | (34–38,41,43,44) | ||||
| NA | A(N1,N2) | 0 | 294 | N | S | X | (33) | |||||
| NA | A(N2), B | 2 | 292 | R | K | X | X | X | (39,42,43) | |||
| NA | B | 0 | 152 | R | K | X | X | X | (36–38,42,43) | |||
| NA | B | 3 | 198 | D | N/E | X | X | (38,43,44) | ||||
| NA | B | 1 | 222 | I | T | X | X | (44) | ||||
| NA | A(N2) | 6 | 222 | I | V | X | X | (44) | ||||
| M2 | A | 20 | 26 | L | F | X | (46) | |||||
| M2 | A | 130 | 27 | V | A | X | (45,46) | |||||
| M2 | A | 8 | 30 | A | T | X | (18,46) | |||||
| M2 | A | 1433 | 31 | S | N | X | (18,45,46) | |||||
| M2 | A | 0 | 34 | G | E | X | (46) | |||||
| HA | A | 2030 | HA cleavage site | ≥5 R or K | +++ | (24,25) | ||||||
| NP | A | 92 | 319 | N | K | ++ | (31) | |||||
| PB2 | A | 3181 | 627 | E | K | ++ | (29) | |||||
| PB2 | A | 177 | 701 | D | N | ++ | (29,30,31) | |||||
| NS1 | A | 97 | 92 | D | E | + (M) | + | (27,26) | ||||
| NS1 | A | 1240 | 80–84 | XXXXX | – | + (M) | + | (27) | ||||
| PB1-F2 | A | 1416 | 66 | N | S | ++ | (28) | |||||
| Segment | Type (subtype) | Number of isolates | Position (H3 and N2 numbering) | Wild | Mutant | Antiviral resistance | High pathogenicity | Hum. adapt. | Ref. | |||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Neuraminidase inhibitors | Adam. | |||||||||||
| Ose. | Zan. | Per. | ||||||||||
| NA | A(N2), B | 12 | 119 | E | V | X | (38,40,42,43) | |||||
| NA | A(N2), B | 1 | 119 | E | A | X | X | (42,43) | ||||
| NA | A(N2), B | 3 | 119 | E | G | X | (42,43) | |||||
| NA | A(N2), B | 7 | 119 | E | D | X | X | (42,43) | ||||
| NA | A(N1) | 415 | 274 | H | Y | X | X | (34–38,41,43,44) | ||||
| NA | A(N1,N2) | 0 | 294 | N | S | X | (33) | |||||
| NA | A(N2), B | 2 | 292 | R | K | X | X | X | (39,42,43) | |||
| NA | B | 0 | 152 | R | K | X | X | X | (36–38,42,43) | |||
| NA | B | 3 | 198 | D | N/E | X | X | (38,43,44) | ||||
| NA | B | 1 | 222 | I | T | X | X | (44) | ||||
| NA | A(N2) | 6 | 222 | I | V | X | X | (44) | ||||
| M2 | A | 20 | 26 | L | F | X | (46) | |||||
| M2 | A | 130 | 27 | V | A | X | (45,46) | |||||
| M2 | A | 8 | 30 | A | T | X | (18,46) | |||||
| M2 | A | 1433 | 31 | S | N | X | (18,45,46) | |||||
| M2 | A | 0 | 34 | G | E | X | (46) | |||||
| HA | A | 2030 | HA cleavage site | ≥5 R or K | +++ | (24,25) | ||||||
| NP | A | 92 | 319 | N | K | ++ | (31) | |||||
| PB2 | A | 3181 | 627 | E | K | ++ | (29) | |||||
| PB2 | A | 177 | 701 | D | N | ++ | (29,30,31) | |||||
| NS1 | A | 97 | 92 | D | E | + (M) | + | (27,26) | ||||
| NS1 | A | 1240 | 80–84 | XXXXX | – | + (M) | + | (27) | ||||
| PB1-F2 | A | 1416 | 66 | N | S | ++ | (28) | |||||
Ose., oseltamivir; Zan., zanamivir; Per., peramivir; Adam., adamantanes; Hum. adapt., human adaptation; Ref. bibliographic reference number; High pathogenicity could contribute, alone or in association with other mutations, to a higher pathogenicity in avians and/or mammals; +++, high level of confidence; ++, medium level of confidence; +, low level of confidence, or in correlation with other mutations; (M), mammals only.
Browse
The main part of ‘Browse’ interface is depicted in Figure 2. Newly deposited isolates are accessible in the ‘What’s new’ section (Figure 2A), which also contains announcements about OpenFluDB. A ‘quick search’ field (Figure 2B) facilitates the finding of new isolates based on their EPI_ISL_ID or EPIID, or isolate name or DDBJ/EMBL/GenBank sequence accession number. The basic ‘browse’ form (Figure 2C) comprises several multiple select menus to restrict the search on virus type, subtype, lineage, host and sample collection geographical location. To further restrict the search criteria, additional filters such as those listed in Figure 2D caption can be applied. An estimation of the number of isolates and sequences returned by a query is updated dynamically and displayed when filters are set.
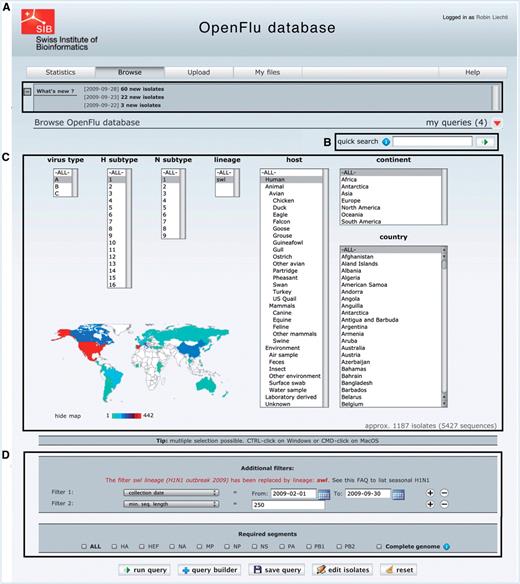
The ‘browse’ form is the major entry point to query the database. (A) ‘What’s new’ section contains links to newly deposited sequences and announcements about OpenFluDB feature development. (B) The ‘Quick search’ field is used to query on EPI_ISL_ID, EPIID, isolate name or EMBL/DDBJ/GenBank accession numbers. (C) Basic ‘browse’ fields are composed of viral type and subtype, viral host and sample collection geographical location. Multiple selections are possible. (D) Several additional filters can be appended to the query; sample collection date, submission date, minimal sequence length, isolate name, EPI_ISL_ID, passage history, lineage, EPIID, DDBJ/EMBL/GenBank accession number, sequence submitter laboratory, whether the isolate has been primarily deposited in OpenFluDB, whether the isolate is publicly accessible in DDBJ/EMBL/GenBank and whether the sequence has a complete CDS. Finally, required segments or complete genome can be specified.
Combined queries, saved queries and alerts. Search criteria selected within the basic ‘browse’ form and the additional filters can be used directly to perform a simple query, or pushed in the query builder tool for further combination with one or several different simple queries. The combinational operator can be intersection, union or exclusion. Both simple and combined queries can be saved for later use as online queries or as daily email alerts containing the list of new isolates and sequences matching the search criteria.
Isolate details and genome annotations. The result of a ‘browse’ action is presented in a tabular form, in which each row represents an isolate and each column represents either an annotation of the isolate or the sequence length of the different genomic segments (Figure 3A). Annotations include isolate name, subtype and/or lineage, passage history, year of collection, submission date, host and country of collection. Columns can be sorted in ascending or descending order, and a filter field enables to display a subset of the results. By default, only nucleotide sequences are presented, but the protein view is accessible using a simple switch at the top of the page. Clicking on an isolate name presents detailed description of this isolate (Figure 3B). This report is split into three parts: the first one containing general annotation on the isolate, mainly similar to the tabular presentation; the second one displaying OpenFluDB-specific annotations; and the third one listing nucleotide or protein sequences depending on the previously selected view. OpenFluDB-specific annotation is a combination of user-provided clinical data, such as host properties and sample collection details, epidemiological data, such as antiviral resistance and protein automatic annotation including antiviral resistance, high pathogenicity and human adaptation motifs.
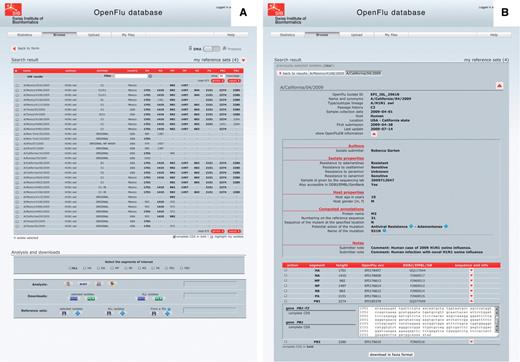
‘Browse’ results are presented in an isolate-centric view. Columns contain either annotations or sequence length. Switching between the nucleotide and protein views is done with the top right switch. Complete CDS or proteins can be highlighted in bold. Checkboxes are used to select isolates for further analysis, export or assignment to a reference set. Analysis buttons are from left to right: MUSCLE MSA, BLAST, SSMs and isolate geolocation. Isolate annotations are exported in a Microsoft Excel file and viral nucleotide or protein sequences are exported in a FASTA file.
Reference sets. Virologists tend to keep a set of reference isolates (also known as clade-specific isolates), option is therefore given to save several lists of isolates, and use them later to compare new query results with these references.
Export. The result of a search query can be exported as a pair of one Microsoft Excel file containing isolate annotation and one FASTA file containing nucleotide or protein sequences. The user can define a set of annotation fields to export, which is saved as a preferred set and presented by default in the next export events.
Analysis
OpenFluDB offers several analysis tools to mine its genetic and epidemiological content. These tools are applied on a selected set of isolates resulting from a search query.
BLAST. This tool presents a list of isolates having the most similar sequences to a user-defined query sequence. To optimize the speed of the ‘BLAST’ interface, blast scores are pre-computed for all sequences against all sequences within each viral segment using an implementation of the algorithm for the Oracle database management system (47). To depict the relation between sequence similarity and sample collection geographical location, the isolates returned from a BLAST analysis can be positioned on a world geographical map. By restricting the similarity threshold, sequences below the specified value are hidden on the map and in the BLAST result table.
Geolocation. The Google maps API (48) combined with geographical data from GeoNames (14) are used to position sample collection location on a world map. Four levels of details from country to administrative division (state) to administrative subdivision (county) to precise geoplaces (cities or village) are represented as different color marks on the map. Clicking on one of these mark displays a pop-up balloon listing the represented isolates with links to their detailed description (Figure 4A).

OpenFluDB analysis tools. (A) The Google Maps API is used to position sample collection on a world map. MSA are computed with MUSCLE and displayed with Jalview (B) and as a tree with PhyloWidget (C). SSMs of hemaglutinin nucleotide sequences are computed by multidimensional scaling, and projections on the first two principal dimensions are displayed. (D) Human H3N2 sequences are colored based on year of sample collection revealing a genetic drift of the sequences. (E) H1 sequences are colored by host species. Three main clusters are revealed: human, swine and avian. Red highlighted sequences from the recent human swine H1N1 lineage are located near the swine cluster.
A similar representation is available in the ‘Statistics’ section of OpenFluDB. By default, all isolates are displayed on the map, but filters on type/subtype, host, continent and country can be applied to restrict the view. At low magnification, isolates are grouped by continents. As zoom level is increased, isolates get grouped by countries, administrative divisions and subdivisions, thus generating more detailed maps.
MSA and phylogenetic tree. Multiple alignments of nucleotide or protein sequences using the MUSCLE program (49) can be provided. To start an alignment procedure, at least two isolates and one segment must be selected from a ‘Browse’ result table. If multiple segments are selected, one alignment will be produced for each of the segment. The alignment can be visualized with the help of the Jalview applet (50). Jalview has been modified to format the alignment in a virologist-friendly way. A consensus sequence is presented at the bottom of the alignment, and nucleotides or amino acids similar to the consensus are presented by dots whereas gaps are symbolized by dashes (Figure 4B). A printer-friendly PDF version of the alignment can be produced. The alignment tree computed by Muscle can be visualized and edited with the help of the PhyloWidget applet (51). Each leaf of the tree contains links to the detailed view of the referred isolate (Figure 4C). This applet can also produce a printer-friendly PDF version of the tree. The corresponding tree can be exported as a text file in standard ‘dnd’ format, whereas the alignment can be exported in ‘ClustalW’ format.
SSMs. SSM is a phylogenetic tool that lets users explore the evolutionary relatedness among influenza virus isolates, mapping several features such as year of collection, hosts or geographical location. These maps enable the study of a large number of isolates at once in a user-friendly interface, to the contrary of classical phylogenetic trees that tend to be difficult to analyze when dealing with thousands of sequences. These maps are produced by applying multidimensional scaling algorithm (52–54) on sequence pairwise distances to transform and project sequence positions into three dimensions. The distances are calculated starting from raw BLAST scores. There are several maps calculated for various nucleotide and protein sequence subsets. The most comprehensive maps present ‘landscapes’ of each of the eight RNA segments and each of the 13 corresponding proteins. Additionally, detailed maps are calculated for each of the 16 hemaglutinin gene subtypes and 9 neuraminidase gene subtypes of type A. All RNA and protein maps are separately computed for both all-species and human isolate subsets. Human maps are supplemented with ‘landscapes’ of the epidemiologically important H1N1, H1N2, H3N2 (Figure 4D) and H5N1 type A human cases. In total, 110 maps are recalculated on a daily basis and presented either under the ‘Statistics’ section or as follow-up analysis from a ‘Browse’ result page. The isolates selected from ‘Browse’ are highlighted on similarity maps. It is possible to zoom into a particular locus of the map to visualize dense areas in more details. Additionally, a subset of sequences/isolates selected on SSM can be sent to the ‘Browse’ interface to access their complete records. To help discover trends and identify outliers, epidemiological data such as year of sample collection, virus type or host species can be overlaid using color gradients. By default, projection on the first and second scaling dimensions is displayed, but all three projections are available to help resolving clusters of sequences eventually obscuring each other.
Case study: H1N1 swl
To better illustrate the main features of OpenFluDB, let us assume that a user wants to find out all isolates of the newly appeared swine-like (swl) H1N1 human lineage, retrieve HA sequences, and then use them to evaluate relations between the H1N1 swl and the seasonal H1N1 strains by means of both MSA (MUSCLE) and SSMs.
From the ‘Browse’ section, set basic filters to select isolates from type A, subtype H1N1, lineage swl, isolated on human hosts. The 2009 A/(H1N1) flu outbreak started in April, so we set a ‘collection date’ additional filter with ‘April 1st 2009’ as ‘From’ value. Finally, one has to select HA as a ‘required segment’. Clicking on the run query button will launch the search procedure. A few hundred isolates are listed sorted by submission date in descending order. To calculate a MSA from these isolates, all the isolates must be selected as well as HA segment in the ‘required segment’ section. Clicking on the ‘sequence alignment’ button will launch the computation. Once finished, the alignment is visualized in Jalview or as a phylogenetic tree by clicking on the corresponding buttons. Both views can be saved in PDF format. Although MSA does show all substitutions and insertions/deletions pedantically, a ‘bird’s eye view’ is a convenient way to globally see these differences and thus relate sequences to each other. For that, the HA sequences from the swine flu outbreak can be compared to other HA sequences from A/H1 virus types on a similarity map by clicking on the ‘similarity map’ button of the ‘analysis’ section of the ‘Browse’ result page. By default, the map of human H1 sequences is displayed, however, since this lineage is a reassortant, it is more interesting to compare it to viruses infecting animal hosts. To do so, we select ‘all’ in the organism filter. Selected sequences are highlighted as big red discs. The map is composed of three main clusters of sequences (Figure 4E). By selecting ‘host’ in the ‘color code annotation’ annotation field, one can observe that one cluster is mainly composed of viral sequences isolated from swine, another one is mainly composed of viral sequences isolated from human and a third one mainly composed of viral sequences isolated from avian species. Interestingly and as described by Garten et al. and Smith et al. (7,8), all highlighted human H1N1 swine-like HA sequences are aggregated in the ‘swine’ cluster.
Implementation
OpenFluDB is implemented as a quite complex, but robust combination of modules written in several programming languages. The database foundation, Oracle DBMS, was chosen because of its recognized data storage reliability, security and rich programming environment necessary to handle complex biological data types. Both Java and PL/SQL are used to implement background algorithms such as isolate annotation and import/export of isolate records from/to GenBank. The OpenFluDB user web interface is developed in PHP with systematic use of AJAX technique.
Conclusion
OpenFluDB provides a convenient and reliable mechanism to collect, manage, store and distribute worldwide influenza data. The tight links between the database, the SSM and the isolate geolocation tool are innovative functionalities to mine Influenza genetic and epidemiological data as well as its evolution. Constant surveillance on the contents of OpenFluDB by means of manual and automatic curation ensures its high reliability. Inconsistencies are reported to the users who are in turn encouraged to report unavoidable inaccuracies or missing features. Future improvements of OpenFluDB will contain pre-computed global MSAs and will offer the possibility to perform ‘robust’ phylogenetic analysis (e.g. Beast). It is our hope that this database becomes an important scientific tool to the whole influenza community, and it has already been successfully used in the early alerting of the novel A/(H1N1) pandemic case by providing access to the sequence data within a few days (7).
Funding
Part of this work has been financed by Oracle Corporation. The Swiss-Prot group and Vital-IT group are part of the Swiss Institute of Bioinformatics (SIB), their activities are supported by the Swiss Federal Government through the Federal Office of Education and Science. Funding for open access charge: The Swiss Federal Government through the Federal Office of Education and Science.
Conflict of interest statement. None declared.
Acknowledgements
A special thank to Rebecca Garten and Catherine Smith from CDC Atlanta for continuous and valuable comments on OpenFluDB user interface and content. We also thank Naomie Komadina from Influenza Center Australia and Isabella Monne from Institute Zooprofilatico Venezia for their feedbacks and Ron Appel for careful reading of the manuscript. Finally, we would like to acknowledge the group of Geoff Barton (University of Dundee) for their support on Jalview improvement, Linda Yankie and col. at NCBI-Bethesda for the implementation of OpenFluDB structured comments in GenBank entries and Oracle Corporation for technical support for part of this project.
CDC Atlanta (Rebecca Garten, Catherine Smith and Nancy J. Cox); NIMR UK (Yipu Lin, Vicky Gregory, Rod Daniels and Alan Hay); Influenza Center Australia (Naomie Komadina and Anne Kelso); NIH Japan (Tsutomu Kageyama, Takato Odagiri and Makoto Ujike); Veterinary Laboratory Agency UK (Bhudipa Choudhury and Keith Hamilton); Food Agricultural Organization (Mia Kim and Gwenaelle Dauphin); Institute Zooprofilatico Venezia (Isabella Monne, Alice Fusaro and Ilaria Capua); WHO Headquarters Geneva (Wenqing Zhang, Katarina Prosenc, Elizabeth Mumford and Keji Fukuda); NCBI-Bethesda (Linda Yankie and David Lipman); Swiss Institute of Bioinformatics (Ron Appel); and Vital-IT group (Roberto Fabbretti, Volker Flegel, Olivier Martin and Sebastien Moretti).
References
Author notes
The authors wish it to be known that, in their opinion, the first three authors should be regarded as joint First Authors.

{kind=link}
{kind=link}
{kind=link}
{kind=link}