





Abstract
The model organism Encyclopedia of DNA Elements (modENCODE) project is a National Human Genome Research Institute (NHGRI) initiative designed to characterize the genomes of Drosophila melanogaster and Caenorhabditis elegans. A Data Coordination Center (DCC) was created to collect, store and catalog modENCODE data. An effective DCC must gather, organize and provide all primary, interpreted and analyzed data, and ensure the community is supplied with the knowledge of the experimental conditions, protocols and verification checks used to generate each primary data set. We present here the design principles of the modENCODE DCC, and describe the ramifications of collecting thorough and deep metadata for describing experiments, including the use of a wiki for capturing protocol and reagent information, and the BIR-TAB specification for linking biological samples to experimental results. modENCODE data can be found at http://www.modencode.org.
Database URL:http://www.modencode.org.
Background
Since the Human Genome Project concluded in 2003, international funding agencies, particularly the National Institutes of Health (NIH), have continued to focus on large-scale, community resource projects such as HapMap (1), 1000 genomes (2), the ENCODE pilot (3) and many others. Included in this effort are model organism-specific projects, beginning with the sequence of the first multicellular organism, Caenorhabditis elegans, published in 1998 (4), which was quickly followed by Drosophila melanogaster in 2000 (5). Ultimately, the aim of all such large-scale projects is to provide resources for the greater research community. These projects almost always require a centralized Data Collection Center (DCC) where the entirety of the data is integrated, undergoes quality control checks and is distributed to the community with sufficient experimental detail to be clear and useful.
The nature and composition of each large-scale project imposes considerations that affect the data collection strategy employed by any particular DCC. Three major influences are the number of contributing laboratories, their geographic distribution and the number of different data types and protocols involved. The number of contributing laboratories may vary from a handful [the Drosophila genome primarily involved three labs (5)] to dozens (e.g. The Cancer Genome Atlas Project; http://cancergenome.nih.gov/wwd/program). In addition, geography can impose network bandwidth constraints for transferring and locating data, and time zone differences may constrain communications between groups. Furthermore, the data types generated may be homogeneous (e.g. HapMap produced SNPs using a limited number of protocols) or highly variable (e.g. ENCODE is using an eclectic assortment of assays to identify many different genomic features). In all cases, a project’s DCC must handle large quantities of data, ranging from a few hundred gigabytes to petabytes.
The model organism Encyclopedia of DNA Elements (modENCODE) initiative is designed to characterize the genomes of D. melanogaster and C. elegans. As a resource, modENCODE serves the model organism research communities, and complements the related human ENCODE project (http://www.genome.gov/10005107), with the ultimate objective of advancing comparative genomics. The consortium comprises 11 research projects: 4 projects for worm, 6 for fly and 1 contributing to both organisms. The modENCODE project was initially funded for 4 years, but has since been extended to 5 years. Of the approximately $17.5 M annual budget (excluding supplemental funding), 55% supports D. melanogaster efforts, 30% supports C. elegans efforts and the remaining 15% is split equally between the DCC and the Data Analysis Center (DAC). These projects represent 52 different data production laboratories at 33 different research institutions in the USA, Canada and the UK. Even within the DCC, with three contributing institutions, geographic location is a consideration. The DCC principal investigator and three staff members (data liaisons and GBrowse development) are located in Toronto, Canada; one co-PI and four staff members (pipeline, data integration and liaisons) are in Berkeley, California; and a second co-PI and three staff members (modMine) are in Cambridge, UK. This DCC staff is charged with tracking, integrating and promptly making available to the research community all modENCODE data generated for the two organisms being studied. The worm and fly genomes are only 97 and 165 million base pairs, respectively, and are small in comparison to the human genome and the data likely to be produced from the 1000 Genomes or cancer genome projects. Thus, by volume, modENCODE is considered a medium-sized (10 terabyte) project.
Of the three factors described above, the most significant challenge for ENCODE and modENCODE is the diversity of feature types coming from the participating laboratories [e.g. transcription factor (TF) binding site characterization, mRNA transcription levels, ncRNAs, stage-specific gene models, chromatin states and DNA replication control], multiplied by the use of a wide variety of different methods and platforms. This is further complicated for the modENCODE DCC by the need to accommodate and integrate data from two organisms. In addition, each participating laboratory must take advantage of cutting edge technologies, and consequently, data production often pushes the envelope of contemporary data storage capacity, requiring a DCC to keep pace.
The metadata challenge
In the context of these operational requirements, the modENCODE DCC’s overarching objective is to ensure that the community is provided with knowledge of the experimental conditions, protocols and verification checks used to generate each data set so that the corpus can be effectively used in future research. Perhaps the greatest challenge in making the large and diverse body of data available to the greater community is providing easy lookup of relevant submissions. Beyond a basic species-specific query, the type of questions that we want the community to be able to ask include: ‘What submissions use the Oregon-R strain?’, ‘Which transcription factor antibodies were produced in a rabbit host?’, ‘Find only those experiments where worms were grown at 23°C’, ‘Find the genomic regions expressed only during pupal stages’, etc. However, an interface is only useful if queries return all relevant results. The factors most critical to making such queries possible are uniformity in data representation, and the completeness and specificity of the associated metadata.
Metadata standards have long been recognized for their utility in making experiments more understandable and integrative. For example, Minimum Information About a Microarray Experiment (MIAME) in conjunction with the Microarray and Gene Expression Data (MGED) ontology has become the standard for describing microarray experiments in the major data repositories, including Gene Expression Omnibus (GEO), ArrayExpress (AE), Short-Read Archive (SRA) and the National Center for Biotechnology Information (NCBI) (6). However, despite the existence of a standard ontology, each repository has its own level of ‘control’ that it imposes on its MIAME-compliant data. AE takes a more controlled approach to collecting metadata, and many of the required MIAME items are specified through controlled vocabulary (CV) terms from the MGED ontology (7). NCBI, on the other hand, has taken a looser approach; its MIAME metadata is collected in free-text form. The benefits to a more controlled approach are that the resulting metadata is more uniform and more amenable to computational reasoning. The drawback is that it may not be quick and easy to specify the metadata since many biologists are unfamiliar with the CVs or ontologies used. NCBI’s approach presents a much lower barrier to entry, which they suggest encourages a high rate of deposition (8); however, the freedom of expressivity that comes with free text has consequences in less-consistent, and often underspecified, descriptions of the experimental details (9).
With the success of MIAME, there followed many additional ‘Minimum Information’ standards groups, collected together under the umbrella of Minimum Information for Biological and Biomedical Investigations (MIBBI) Foundry (10). Of particular relevance is the draft of the Minimum INformation about a high-throughput SEQuencing Experiment (MINSEQE) (http://www.mged.org/minseqe/), although this proposal is still in draft form and does not yet have a concrete specification.
The NGS challenge
The modENCODE DCC's efforts to standardize its metadata collection was complicated by the rapid shift to next-generation sequencing (NGS) that occurred just as the project was getting underway. At the beginning of the modENCODE project, NGS throughput had begun an exponential rise that continues to this day, but GEO was only just starting to accept short-read data and the SRA was not yet up and running. Anticipating the change in technology usage, the modENCODE DCC began preparing to accept and process high-throughput NGS data. To this end, we created a concrete realization of the MINSEQE standards for the modENCODE project.
From discussions with the ENCODE group and the experiences reported by AE, we knew that collecting metadata would be one of the largest challenges we faced. To support the types of queries mentioned above, the modENCODE DCC devoted considerable time and attention to the metadata collection process. It would require active collaboration with the data providers by biologically trained staff knowledgeable in the experimental techniques, data types, data formats and software that would be employed. Additionally, we knew the volume of data submitted would necessitate scalability and as much automation in the data quality control process as possible, yet the diversity of experiment and data types would require flexibility and swift responses to changing requirements, two demands, which are often incompatible.
An effective consortium DCC must make a large volume of data readily accessible to the research community as soon as the data are experimentally verified. To respect the research objectives of DCC data producers, resource users are encouraged to observe a 9-month waiting period. During this time, they may freely use the modENCODE data in their own research programs, but must defer publications until either after the waiting period or until they have conferred and obtained agreement from the original producers. (The modENCODE data release policy is available at http://www.genome.gov/27528022). We present here several principles in the design of the modENCODE DCC and our approach to collecting, storing and cataloging data. We describe the ramifications of collecting thorough and deep metadata for describing experiments. The lessons we have learned are applicable to both large data centers and small groups looking to host data for the broader community.
Results
The primary DCC mandate is to provide a research resource for the greater community. Just as people searching the web seldom look beyond the first one or two pages of results, researchers cannot be expected to find what they are looking for simply by browsing through a catalog with thousands of entries. The usability of such a large resource is dependent on its ability to catalog, categorize and query its contents using those indices reflective of key experimental variables so that users may clearly narrow their searches to the most pertinent results. Indeed, for production tracking purposes the NIH required a report from the DCC listing how many data sets had been produced for each of the different experimental types, developmental stages, tissues and so forth: something that is only possible if this information is captured at its origin.
To accomplish this, the DCC needed to collect the experimental details describing the biological sample, protocols, reagents, parameters and other information associated with each data set. Ideally this metadata should be of sufficient detail that it would be possible for another scientist to fully understand and repeat that experiment. We employed a combined approach to accomplish this, using both free text and CVs. Data providers detailed their experiments as thoroughly as possible with free text, and key experimental factors, such as cell type or tissue, developmental stage and so forth, were specified using CV and ontology terms to facilitate categorizing, querying, downstream integration and analysis of the data from these diverse experimental approaches. A corollary requirement followed from this; the need to track the relationship between the original biological samples and experimental protocols to the resulting raw data and derived annotations. This requirement was met by extending the ArrayExpress MAGE-TAB metadata format [originally developed for microarray data to connect samples to the resulting data (11)], to a format called BIR-TAB (Biological Investigation Reporting Tab-delimited) that is flexible enough to handle the variety of experiment types that were required. The third requirement did not concern functionality, but rather the timeline, the DCC needed to be operational and ready to receive data within the first 6 months because the data production laboratories began generating data immediately upon funding. To meet this deadline, we simplified the process by restricting the collection of both raw and analyzed data to a small number of standardized formats, such as WIG and GFF3. Furthermore, we took advantage of existing open-source software components whenever possible in order to speed development.
The completed DCC pipeline for processing modENCODE data sets is a multistep procedure. As illustrated in Figure 1, and detailed in the following sections, the process begins with discussions between an experimental lab and a DCC curator. These discussions lead to an agreement on what metadata is sufficient to fully describe an experimental run, including the type and format of raw data files that are submitted in association with that run. Once the prepared metadata is completed and raw data are submitted to the DCC, the complete package is run through a series of automated checks, followed by additional manual quality control (QC) by DCC curators. After the submission passes both QC steps, the data submitter gives final approval for release, at which point the DCC makes the data available to the community to browse, search and download. The data are also released to larger public repositories including the model organism databases FlyBase (FB) and WormBase (WB) and GEO.
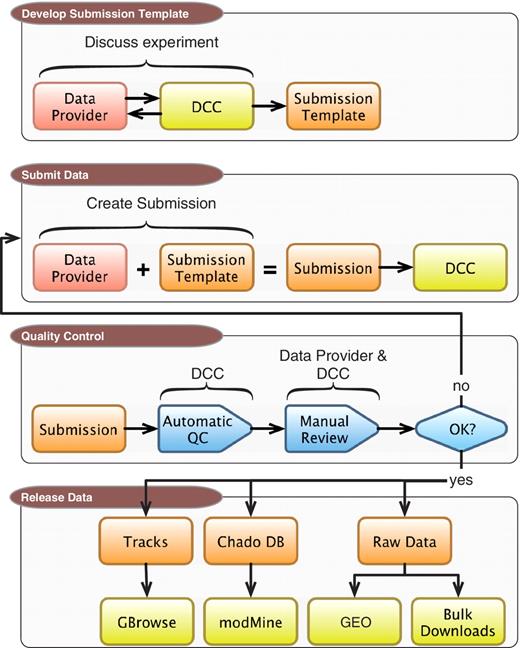
DCC workflow. Submitting data to the modENCODE DCC can be divided into four parts. It begins with discussions between a data provider and a DCC curator to determine the required metadata and data formats for a given category of submission. Once the submission template is made, the data provider can prepare and submit a data set to the DCC. The data set undergoes a series of automated and manual QC checks. If the submission does not pass these steps, it is returned to the data provider and/or the DCC curator for modification. Once a submission satisfies all requirements, and is approved by the DCC and data submitter, it is distributed to the community through the GBrowse genome browser, modMine query interface, graphical submission filtering tool and the public repositories of FB, WB and GEO.
Figure 2 illustrates a simplified model experiment submitted to the modENCODE DCC, which we will reference throughout this article to describe the different components of our system. It shows a typical sequence of experimental steps for a ChIP-seq experiment, from the worm culturing through chromatin extraction, sequencing and analysis. The DCC defines an individual submission as a single experimental factor (such as a TF) tested in a single developmental stage, cell line or tissue, together with its controls and replicates (a minimum of two are required). Each submission is part of a larger collection of experiments that employs the same assays to test a variety of factors in a variety of conditions. Figure 2 also shows the different components we use to collect the experimental details for the model experiment, as discussed in the following sections.
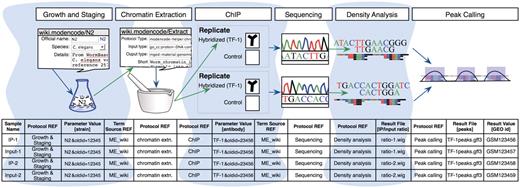
A model experiment submitted to the modENCODE DCC and its mapping to metadata components BIR-TAB SDRF and the wiki. The top half is a diagram of experimental steps for a model ChIP-seq experiment: a worm culture is prepared, the genomic DNA associated with chromatin is extracted, followed by division of the extraction into two biological replicates. These are further subdivided, with half of each DNA sample used as a control, while the other is exposed to a specific TF antibody in a ChIP step. The resulting materials are prepared for sequencing, and the data processed to identify the set of binding sites occupied by the TF tested. The corresponding BIR-TAB SDRF is shown in the bottom half, and mirrors the flow of experimental steps as indicated by the green (output) and blue (input) arrows. The inputs and outputs are the arcs connecting each protocol node of an experiment represented in the database. Each cell in a protocol column of the BIR-TAB file maps to a specific wiki page where the inputs and outputs of that protocol have been indicated. Most experimental parameters, such as strain and antibody, are also specified in the wiki. A reference to the wiki for these experimental parameters or results is indicated with a Term Source REF column immediately following the parameter column.
Acquiring thorough experimental details from modENCODE data providers
The volume of data produced by the modENCODE consortium is sizable, easily two orders of magnitude larger than FB or WB at the beginning of the project. Since the number of different data sets produced by modENCODE would be very large, it is impractical to list them individually as tracks in a browser. Additionally, we knew end-users would require more than the lists of precategorized data; they would need the flexibility to query data sets using a range of different experimental factors to locate the precise data sets applicable to their own research.
Using a wiki to collect experimental metadata
Collecting a large amount of descriptive data in a controlled way requires a user interface for entering this information that is aware of the pertinent CV for different fields. Additionally, because of the geographically distributed nature of the project, a browser-based interface would be most convenient for users. Given these constraints, the only practical approaches we could use were either HTML forms or a wiki. Given the timeline and the need for rapid deployment, we chose to use a wiki for speed of implementation, presumed familiarity and ease of use by the consortium, the ability to handle both free-text and related images and support for extensions that would allow us to add forms for structured data.
The modENCODE wiki (http://wiki.modencode.org) uses MediaWiki software, with an additional plug-in developed by the DCC. Our DBFields extension allows wiki editors (generally DCC staff) to use HTML-like syntax to create a form on any wiki page with fields that can be free entry, selection boxes or auto-completing text fields when entering CV terms. In addition to enforcing the CV, any of these fields can be marked as ‘required’ so that, for instance, a protocol will be marked incomplete until an assay type is provided. Every change to a MediaWiki page generates a new unique URL for that version, and the DBFields extension is integrated with MediaWiki's versioning system so that changes to the form contents are also tracked. An example of a DBFields-templated wiki page is shown in Figure 3.
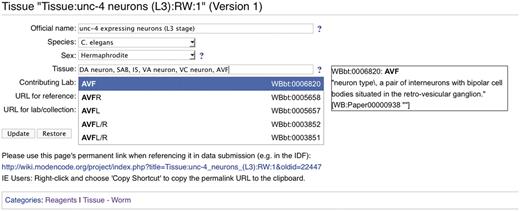
Screenshot of a modENCODE wiki tissue page using DBFields template. In this example, WormBase cell and anatomy ontology (24) terms are selected to describe unc-4 expressing neurons in the L3 stage. The DBFields template for tissues was configured to include fields for a colloquial name, species, sex, tissue, contributing lab and related external URLs. The tissue field allows for multiple selections from the configured ontology; as the user starts to type a phrase (such as AVF), partial matches are displayed for selection and the corresponding definition is displayed on the right. After the user ‘Updates’ the form to accept the changes, an updated URL is displayed for the user to refer specifically to this version of the wiki page. This URL is used in the BIR-TAB metadata documents to describe the sample, and the vetting software retrieves the field values during processing.
The wiki is divided into three basic categories for collecting experimental metadata in a controlled way: experimental descriptions, protocols and reagents. Each of these wiki categories uses a DBFields extension template to record the specific attributes required. When a laboratory submits data to the DCC, our automated pipeline refers to the appropriate wiki page to check individual fields in each category and retrieves their values as required by the pipeline’s different software modules. Upon release of a submission by the DCC, all referenced wiki pages become public and available to the community. Of the 1112 released submissions to date, there are references to 54 experiment descriptions, 399 protocol descriptions and 600 reagent descriptions, totaling 1049 unique wiki pages (Figure 4A).
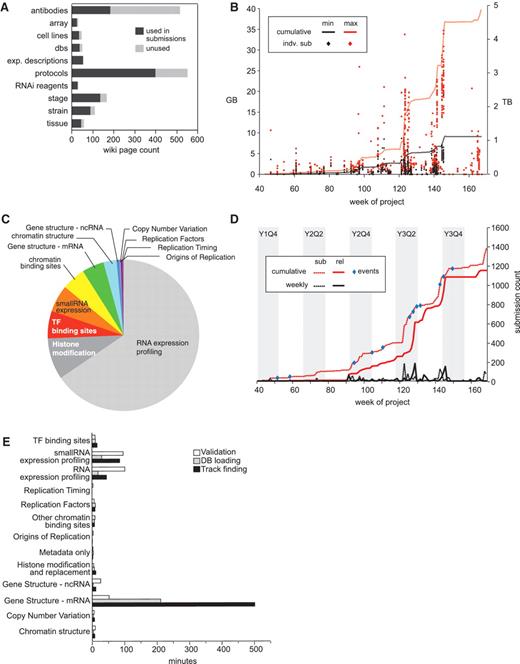
modENCODE data submission statistics. (A) Distribution of wiki page types. Number of wiki pages used in released submissions (dark gray) out of the total set, which have been entered in the wiki. The unused set of wiki pages may be used in future submissions. Data were only from released data sets, and not those superseded, deprecated or rejected. (B) Distribution of submission package sizes. Scatterplot of individual package sizes (in GB, scale on left) are overlaid with the cumulative size of all modENCODE data (in TB, scale on right), over the course of the project. Black indicates the size of the files uploaded into the system by data providers, and is the minimal set required for backup; red indicates the total size of a processed submission, including gbrowse tracks, chadoxml and all versions of uploaded data, and is the maximum size required to maintain a complete history. (C) Composition of modENCODE data types. These are based on the cumulative submission file sizes in each category, including data sets that have been superseded, replaced and rejected. (D) Number of submissions over time. Plot reveals spikes in data submission. Dotted lines indicate when submissions were initially created; solid lines indicate when submissions were released in the pipeline. Red lines show cumulative counts; black lines show the number of counts per week. Events, such as scientific meetings or data freezes are indicated with blue circles. Project quarters are indicated (Year 1 Quarter 4 is abbreviated Y1Q4). All data, including superseded, replaced and rejected submissions, are shown. (E) Pipeline processing times grouped by data type. Average processing times (in minutes) for the three pipeline steps (validation, database loading and track finding) are shown for each type of data in released data sets.
The experimental description wiki pages record a high-level description for each set of experiments carried out by individual laboratories. This description consists of a ‘data type’ tag to broadly classify the genomic features or behaviors being identified, an ‘assay type’ tag to classify the experimental technique applied, and a short paragraph to describe the set of experiments that fall under this umbrella classification. These classification tags are used for reporting by downstream applications. In the example in Figure 2, the submission belongs to a set of ChIP-seq experiments with TF antibodies, and would be tagged with data type ‘TF binding sites’ and assay type ‘ChIP-seq’. To date, we have encountered 23 different submission types, including: ChIP-seq and ChIP-chip investigations of TF or other binding sites, replication timing, histone modification and chromatin structure, gene annotation, 5′- and 3′-RACE, targeted RTPCR, and RNA tiling array and RNA-seq to identify transcription levels.
Protocol descriptions are fundamental to a modENCODE submission, providing the details for each experimental replication and a framework for the key experimental variables. The protocol description pages are as basic as possible to ensure they are appropriately used, requiring only the protocol's type, inputs and outputs of data and reagents used and produced, and a prose description. Although a protocol can be comprehensive enough to describe an entire experiment, we have encouraged data submitters to be granular. For example, an organism growth protocol should be separate from a subsequent chromatin purification protocol (as in Figure 2). A typical experiment will have protocols for organism growth and isolation, sample preparation, library preparation, sequencing/arraying, alignment/normalization and peak calling. This granularity enables data providers to reference the same protocol in different experiments; for example, the same organism growth protocol may be reused regardless of the assay applied.
The reagent category of wiki pages comprises several subcategories. Reagents represent the experimental factors that differ between related submissions. Sub-categories include antibodies, strains, cell lines, developmental stages, tissues, RNAi reagents, microarray chipsets and recombinant constructs/vectors. Each of these types of reagents uses its own form template with fields designated by the DCC curators in cooperation with the data submitters. The largest subcategory is antibodies with over 500 antibodies specified by the consortium (though only approximately 180 of these have been referenced in released submissions to date).
All submissions to the DCC begin with a dialog between the DCC curator and the lab submitting data to determine the protocols, reagents, metadata and data formats appropriate for a given category of experiment. The curator acts as an advocate for the end-user making sure that sufficient informative detail is provided. Because experimental categories vary widely across the modENCODE project, this requires metadata and data design on a per-laboratory basis, although once original templates are produced they can subsequently be followed for similar experiment categories. Based on the discussions with lab personnel, the DCC curator sets up a collection of wiki page ‘stubs’ for the lab to fill in. Upon public release, this metadata is incorporated into our public databases and supports queries in modMINE, is the basis for the generation our matrix-based download interface and provides the track descriptions for GBrowse, a web-based genome viewer.
Linking the wiki and data together with a submission template
When submitting data to modENCODE, data providers need to connect the descriptive wiki pages to the resulting data files. This information is supplied to the DCC in BIR-TAB format via two accompanying metadata documents: an Investigation Design File (IDF) and a Sample Data Relationship File (SDRF).
The primary objective of the IDF is to provide details about the overall experiment, such as a name, data submitter details, protocol references and CV definitions. The BIR-TAB IDF file is nearly identical to the MAGE-TAB IDF file format, with extensions allowing the experiment and protocol details to be indicated via references to the appropriate wiki URLs, the ability to indicate both project PI and individual laboratory co-PIs and an additional ‘format type’ field for indicating the appropriate CV or ontology to use as the source of permissible terms in specific fields. This means that BIR-TAB can support multiple formats for CV or ontology term sources. At present, in addition to the formats already supported in MAGE-TAB, the syntaxes include the OBO format, a MediaWiki URI and intersubmission references for handling replicates and controls.
The BIR-TAB SDRF file links the derived raw and processed data files to original biological samples through a series of protocol steps (Figure 2). Whereas MAGE-TAB SDRF uses a structured format that mirrors the processes of doing a microarray experiment, BIR-TAB SDRF has been generalized to expect an arbitrary series of experimental protocols and their inputs and outputs. These protocols can be any mixture of bench and computational procedures. As the example in Figure 2 illustrates, the SDRF flattens the sequence of experimental steps into a table. Internally, the experiment is stored as a directed acyclic graph (DAG) with protocols and reagents treated as nodes. These nodes are represented as columns in the SDRF. Each protocol maps to a specific wiki page; any sample characteristics or sample-specific treatments such as stage and antibody that are captured in the wiki are also referenced in the SDRF. Using a wiki as the repository for the experimental details reduces the likelihood of inconsistencies in the BIR-TAB files and provides versioning so that changes to protocols and sample descriptions can be tracked over time.
The BIR-TAB files accompany each modENCODE submission, allowing the DCC to track the unique combination of experimental factors and link together the descriptive information for each biological sample with the final raw data and annotations for each submission.
Processing and quality control of modENCODE data
The modENCODE submission pipeline handles the management and tracking of submissions in four automated stages: data upload and expansion, QC, populating the DCC database and browser track generation. Once these steps are complete, the DCC liaison and the data provider sign off on the submission and it moves to public ‘released’ status. The pipeline tracks all uploaded submissions, including the date, result and status reports at each stage of processing. The DCC assumes that the respective data providers have performed biological QC prior to submission, which varies by data type. The DCC is only responsible for verifying what the data providers report, not the quality of the experiments themselves.
Submission and tracking interface
The submission interface is implemented using Ruby on Rails, a web application framework designed for rapid development. We inherited Rails and the skeleton of the submission pipeline from the ENCODE DCC. It has proved very well suited for our needs; in particular, development of new features is very fast, including everything from adding to the data model, to developing new views of the pipeline state.
We initially planned to continue developing the pipeline software in conjunction with the ENCODE-DCC, but further exploration indicated that our respective requirements were too different. Although both DCCs track and manage incoming data, we implemented more automatic processing dependent on CVs, which required a more complex job management system. Developing a working solution was more critical than maintaining a commonly shared generalized solution during the early stages of the project, resulting in the two DCC’s submission pipelines bearing only a superficial resemblance (Figure 5). This experience emphasizes how difficult sharing software across projects continues to be even when the projects are as similar as ENCODE and modENCODE.
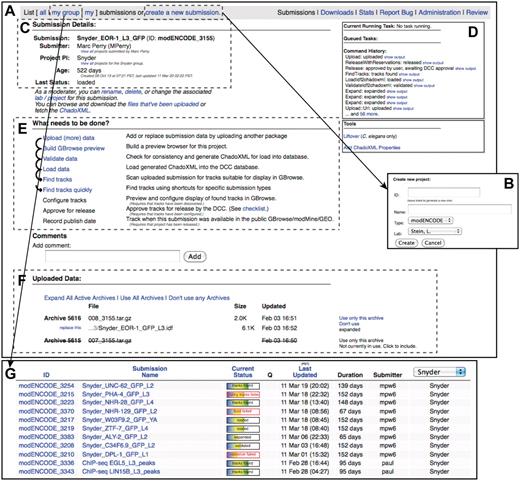
modENCODE submission interface. (A) The primary page for an example individual submission is shown. (B) New submissions are created by entering a name for the submission and selecting the appropriate laboratory and PI. (C) Once a submission is created, the current details are listed on the upper left side of the page. (D) The step-by-step series of tasks that are being executed by the pipeline can be monitored in real time, and the corresponding output from each module can be viewed. (E) Progress is indicated as the submission moves through each step of automated QC processing. In this example, all that remains to be done is configuring the tracks for the browser, final manual checklist and public release. (F) All of the primary files making up the submission package are listed on this page: the IDF, SDRF, wig and GFF3. Individual files may be replaced, if desired, by the submitting laboratory. (G) A list of active submissions can be displayed separately, providing the user with a snapshot of the vetting status of their submissions.
Automated QC
To enforce consistency across all submissions to the modENCODE DCC, we developed a modular automated vetting tool written in Perl. To vet a submission, the tool first scans the BIR-TAB documents. Assuming that there are no syntax errors or technical inconsistencies, the flat metadata is turned into the graph structure representing an experiment. Next, all wiki pages referenced by the submission are fetched from which all field values are collected and used to fill in the metadata. Since the protocols in the wiki contain CVs describing its input and output ‘types’, the consistency of each protocol’s inputs are reconciled with the preceding protocol’s outputs to confirm that the series of protocols making up the experiment graph built from the SDRF matches that in the wiki. If this is confirmed then, based on introspection of the experiment graph, vetting modules are selected for execution.
The vetting modules include simple checks, such as making sure that specified ‘Result Files’ actually exist in the submission data set, as well as more extensive checks such as ensuring that antibodies have had sufficient QC prior to their experimental application. There are modules for ensuring the existence of external gene, transcript, protein and EST identifiers, as well as SRA and GEO accessions. We also permit and check references to remotely hosted raw data files. The set of modules also includes support for vetting of GFF3, WIG, BED and SAM data formats. Although GFF3, BED and WIG are in use by many other data repositories, and several vetting scripts already exist, our formatting requirements are more stringent (particularly for GFF3) and we have developed enhanced modules for these formats (see ‘Methods’ section for formatting requirements). As each piece of data is vetted, the experimental graph is updated. For instance, when the vetting package runs across a node in the graph describing a GFF3 file (such as the peak file in Figure 2), it processes the GFF3 file and attaches new genomic feature nodes to the file node representing the GFF3 file.
Vetting submissions takes anywhere from under 30 seconds to as long as 5 days with an average time of about an hour (Figure 4E). This variance is due to the differences in complexity of the underlying data and varying data size. Approximately one-third of the submissions initially contain some type of error which are largely resolved using two basic approaches: first, many errors can be fixed just by taking a closer, more critical look at the submissions; second, if the errors are not obvious, submissions are re-run with truncated data files; this lets us refine and correct the metadata without the slowness entailed in loading a million features from a GFF file (for example). The worst cases (6/3043) are when there are just one or two errors occurring at the end of the data file (e.g. GFF files where a couple of the features toward the bottom of the file have an end point before the start point—which is invalid.) In these few cases, there is nothing to do other than keep testing the data file; this situation often indicates issues in the file generation by the data provider, which we work with them to correct. Typically, the longest running validations are those of full-genome gene models, while the shortest running are array-based submissions (ChIP-chip or tiling arrays).
Data storage and track generation
There are several existing database schemas for storing genomic feature data, generally tied to different genome browsers. A partial list includes AceDB, the UCSC annotations DB, Ensembl and Chado (12–15). We chose the Chado database to store the modENCODE experimental metadata and genomic features because of familiarity, availability of tools, nominal compatibility with FB and WB and browser neutrality. In addition, it is highly normalized, which reduces redundancy and the potential for internal inconsistency.
Chado's structure allows for the easy addition of extensions in the form of new tables, allowing us to extend the schema to accommodate generic submission details, protocols and data references (Supplementary Figure S1). The new extension takes advantage of existing tables for CV and external database references, and links protocol inputs and outputs to the genomic feature table where appropriate. Since these tables are an extension, they do not interfere with existing tools developed to work with Chado databases.
Following automated vetting, submissions are loaded into the database. Database loading was simplified by generating ChadoXML as an output of the automated vetting process. In situations where the data did not lend themselves to a relational schema, such as ChIP signal data, we kept the data on the file system and recorded links to these external files in the database. The load times varied widely depending on the submission, from 7 s to 2 days, with an average time of 30 min (Figure 4E).
The last step of the automated process is the direct generation of GFF3, WIG and SAM files from our database for display in our public browser and for community member download, thereby ensuring internal consistency across the entire modENCODE project. This added anywhere from 1 s to 12 days to the data processing times, with an average of about an hour. For both loading and track generation, the variability was again mainly due to the complexity of the data. Gene features, with multiple levels of sub-features (transcripts, exons, etc.), took the longest time to load (data not shown).
Manual approval checklist
After a data set has passed automatic vetting and been loaded into the database, the responsible DCC curator administers a final check for errors that can only be detected through human review. While initially performed ad hoc, over time these steps have been formalized into a checklist that is incorporated into the pipeline. Common errors include listing an incorrect antibody as compared to the given experimental title, references to retired wiki protocols, inclusion of an insufficient number of replicates and references to GEO IDs that represent the wrong data set. For example, even if a submission is syntactically correct, distinguishing the submission of biological replicate data from a resubmission is crucial for ensuring that a submission package is complete. In addition to these data integrity crosschecks, the curator reviews the experiment’s prose descriptions for comprehensibility by community members (the full checklist can be viewed in Supplementary Data S2). If a submission does not pass these checks, the data submitter and/or curator must edit the submission and fix the problem. The revisions to the metadata and/or data are then uploaded, and the submission pipeline tracks the revision history.
Liftover of data between genome versions is required if we receive C. elegans data sets in coordinates other than the modENCODE agreed standard (presently WS190). Though the original data files remain available for public download, all released C. elegans data have been processed into WS190 coordinates. Our liftover tool is a Java re-implementation of the WormBase tool (http://wiki.wormbase.org/index.php/Converting_Coordinates_between_releases), extended to accommodate GFF3, WIG, BED and SAM (see ‘Methods’ section). Additional manual steps required of the curator prior to release include editing of generated track prose descriptions and configuration of track appearance in the browser.
Once the submission is approved by the DCC, the data submitter is asked to approve it for release. Until the submission is approved, only the raw submission files provided by the data submitter are available to the public.
Formatting, volume and rate of data submission
In contrast to repositories like GEO or dbEST, which deal with one or a small number of assay types, the DCC accommodates a broad set of biological result types and data formats. The original consortium proposals were largely predicated on using the array-based technology then available, and included commercial and custom arrays on multiple platforms (Nimblegen, Affymetrix and Agilent) for RNA expression profiling and identification of TF and chromatin binding sites using ChIP-chip. For array data, we required raw data files in order to make submissions to GEO on behalf of the data providers. We collected signal intensity plots in BED/WIG, and peak calls in GFF3. To date, 556 released submissions (50%) describe array experiments.
Soon after work commenced, NGS became affordable, and many consortium labs supplemented or switched their approaches to use this newer technology. Due to the large size of sequencing files (FASTQ), we were not able to accept them without a significant investment in hardware and systems management, which would require additional funds and time, in addition to bandwidth constraints. Because our role is primarily to serve as a data coordination center, not a data repository, we instead requested modENCODE data providers to submit their sequences directly to GEO/SRA and then provide an accession number to the DCC. DCC staff then confirms the submissions of raw data. To date, 461 released submissions (41%) describe NGS experiments. This ratio has shifted over time. In the first 2 years of the project, array submissions represented 88%, but that number has shifted to only 37% as NGS becomes more prevalent (data not shown). Figure 4B shows the size distribution of submissions over the lifetime of the project. An overall trend can be observed—more recent submissions are larger than those submitted in the early weeks of the project. This is likely due to the DCC’s requirement that RNA-seq submissions include read alignments in SAM format, also resulting in the bulk of data consisting of RNA-seq submissions (Figure 4C).
Submissions tend to arrive at the DCC in waves prior to events such as scientific meetings, publications and ‘data freezes’. Figure 4D shows the number of experiments deposited in the DCC over the course of the project with the first data sets arriving at the DCC in the fourth-quarter of the first year (Week 40). Enormous increases in data deposition can be observed prior to these major events. In total, we have released 1112 submissions, and processed an additional 83 that have been superseded, deprecated or rejected.
Releasing data to the public
At the conclusion of our vetting process (i.e. approval by the data submitter), a data submission is considered ‘released’. The processed files and related wiki pages are made available via several avenues: immediately from the pipeline ‘list’ interface (http://submit.modencode.org/submit/public/list) or the bulk downloads selection interface (http://submit.modencode.org/submit/bulk_download/); GBrowse for viewing data in the context of the genome (http://modencode.oicr.on.ca/fgb2/gbrowse/worm/ or http://modencode.oicr.on.ca/fgb2/gbrowse/fly/); modMine for querying and downloading of data subsets (http://intermine.modencode.org); and major repositories such as GEO, WB and FB.
The average time between when a data set is marked ‘released’ and its posting on GBrowse is ∼1 week. The ChadoXML is transmitted to modMine for regular public releases on a quarterly cycle. The DCC also submits data files and the appropriate metadata to GEO. To date, we have made 321 full submissions. Additionally, some of the data providers have made their own submissions to GEO. To date, 86% of raw modENCODE data is currently in GEO.
In February 2011, the SRA announced that it was shutting down, which affects the DCC’s data acceptance policy and procedure for NGS data. The DCC has begun to accept sequence files from production labs, and is acquiring existing project data from the SRA, which will be maintained on the University of Illinois at Chicago (UIC) data cloud. This resource is being used for intraproject analysis, and will be made available to the public by the end of the project via Amazon. Since there is an on-going debate within the community over the value of retaining raw data for array and NGS (for example, it is a common practice for commercial sequencing companies to delete files after 6 months), we do not yet have an expiration date for hosting raw data files.
Discussion
Reflections on collecting deep metadata
modENCODE is the first large-scale project for which its DCC collected extremely detailed and controlled protocols and sample descriptions. Our approach allows us to provide complex querying capabilities based on the experimental metadata in our public interfaces (modMine and the graphical submission filtering tool), a feature that tends to be lacking in other systems. For example, it is possible to specifically query the DCC for all ChIP-seq data sets with immunoprecipitated chromatin from 0 to 2 h embryonic flies using antibodies to the CTCF protein, an operation that is currently impossible using the ENCODE browser. We attribute this to our consistent use of CV and ontologies in protocol descriptions and for experimental reagents, combined with thorough review by the curatorial staff. Without this, we would have been limited to free-text queries, and thus unable to provide this functionality to the community.
Using the combined wiki and BIR-TAB metadata approach, we collected unambiguous metadata for 1112 released experiments and connected biological samples to their resulting data and annotations from more than 2700 biological replicates to date. We have been able to accommodate the diverse data and assay types for the project without compromising the depth of experimental details. This flexibility is the direct result of the modularity with which we built the system: the requirements for complex experimental details were not hard coded into our submission pipeline per se, but were dynamically configured in the wiki by curators without requiring re-factoring of the validation code.
However this combination of flexibility to handle a wide variety of experiments, coupled with collecting precise descriptions for each of these experiment types, comes at a cost. The challenge associated with collecting metadata is that it entails time-intensive ‘translation’ by curatorial staff. Preparation of metadata documents for new experiments and protocols is, in essence, the creation of a specification for the pipeline software to interpret. And the responsibility for translating an experimental description, as might be found in a lab notebook, into a machine interpretable form, which would be useful for downstream QC and querying, required meticulous preparation by experienced and well-trained DCC staff.
That said we were able to meet to our goal through other simplifications to the pipeline. At the outset of the project, the standard DCC data formats included WIG, BED and GFF3. With the surge of NGS data, this came to include the SAM format for sequence alignments. Standardized data formats greatly eased the workload on our curators in that custom data conversion was not required. We allowed some flexibility in feature attributes in GFF3 (Column 9), which allowed submitters to include details they felt were important to convey about individual features, such as separate p- and q-values for peak calls, expression levels from RNA-seq and flags to signal whether or not a feature remained predicted or was confirmed. This often made the GFF3 files easier to read and we attempted to make such attributes uniform across the entire project.
The GFF3 format can be used to annotate diverse feature types, and DCC curators were necessarily involved in each new type of feature submission. For groups providing method-specific annotations of gene models and their supportive data, submissions required custom examples of GFF3 files that were developed by DCC staff through email interviews with the data providers. Once the initial file format was finalized, subsequent submissions were made more easily. Additionally, the strictness of the format sometimes illuminated problems in the source data.
Overall, the process of collecting deep metadata was a daunting but productive effort. Significant resources from both the DCC and data providers are needed to ensure that complete and correct experimental details are being collected. While it is possible to collect less specific or free-text metadata, we found that the benefits—detecting errors in data, generating summary reports and supporting complex queries—outweigh the disadvantages—primarily the extra time spent in configuring the information into a machine-interpretable form. The descriptive information we’ve collected allows modENCODE data to be more easily queried and deeply investigated by the scientific community, though its long-term usefulness will only be measured through integration into and use in downstream community portals such as FB and WB. Thus far, the completeness of the metadata has been invaluable in the preparation of the worm and fly integrative analysis papers (16,17), and has allowed the authors to select appropriate data sets for comparative analysis.
Reflections on submission system implementation
The DCC submission system can be divided into four major software components. The wiki to structure and collect experimental metadata, the vetting tool to automatically verify submissions, the Chado database to store genomic features and experimental metadata, and the pipeline interface for uploading, tracking and reviewing submissions.
Wiki
The flexibility of the wiki interface for tracking experimental metadata proved quite sufficient; in addition to supporting formatted text and images, the support for extensions allowed us to develop the DBFields extension and collect important attributes in a structured manner. Furthermore, using the MediaWiki software gave us access to a large number of existing extensions, including a WYSIWYG editor and an interface for marking private pages as public after release.
On the other hand, the loose integration of the wiki(s) and the submission pipeline was a weakness. For instance, accounts on the wiki and submission pipeline are independent, so usernames and passwords can differ. In addition, because many individual laboratories used internal wikis that were not linked to our system, data submitters were entering some of the metadata twice (once in their own private wiki, a second time in the DCC’s wiki). A single consortium-wide wiki might have made this easier, but this would require agreement amongst all data providers, a larger set of resources at the DCC and tighter connections between the DCC and the production laboratories for requirements gathering and implementation. In retrospect, however, we feel a tighter integration between the submission pipeline interface and the wiki would have allowed us to avoid several time-consuming hindrances. Despite these drawbacks, the wiki paradigm enabled the DCC to successfully capture the metadata we set out to. The technology itself has worked well; it supports capturing all of the experimental metadata that we want, and it provides a familiar Wikipedia-like interface for the community to view metadata.
Vetting
The DCC vetting software began life as a Perl script for generating a ChadoXML file from a BIR-TAB/GFF submission, which required a basic level of syntactic validity. We quickly extended its responsibilities to detect logical inconsistencies within a submission, basic checks of accessions and other repetitive tasks that are easier for a computer than a human, and to verbosely report all the error(s) and warning(s).
The vetting tool is designed as a dynamic, modular system. Dynamic, so that submissions can be vetted using only the appropriate modules based on the CV typing of the fields that are unique to that submission. The modularity allowed us to easily and quickly add new modules in response to new data types. The vetting tool builds a full model of the experiment, including all metadata and genomic features, before writing ChadoXML to enable crosschecking of dependent references across fields and features. A drawback of this approach is high memory utilization; keeping track of the full experimental model requires some caching to disk (despite 12 G of available memory), which drastically slows the processing of larger submissions. This is particularly evident in gene model submissions, which have multilevel features (of genes, transcripts, exons), and are 30 times slower processing than the average of other types of submissions.
For most data sets, however, the approach is satisfactory. In particular, new modules can often be developed in a day or two. This short response time has proved critical as the types of data provided and requirements for validation have changed over the course of the project. For future projects looking to do metadata-based verification, we recommend the modular approach, as well as examining new methods that allow distributed processing of different components and avoid the need to examine the entire submission as a whole.
Chado
In practice, we have found Chado to be sufficient for its primary task of storing genomic features, and with our extension to link features to experimental metadata, making it easy to build browser tracks, populate modMine and package GEO submissions by filtering data associated with particular submissions. We found it necessary to partition our main Chado database by creating separate namespaces for each submission. This made it possible to remove or reload unreleased submissions from the database, which are tasks that need to be performed on a regular basis as part of the vetting. Unfortunately, this approach makes queries across all submissions more difficult to write and time consuming to perform. The modMine group mitigates this somewhat by generating a read-only Chado database with submissions partitioned by PI rather than by individual submission, which they use to build the modMine query database.
One of the big limitations of Chado, and indeed, any schema designed solely for genomic features, is the lack of support for continuous data such as signal intensity. Of course, extremely high-density genomic feature data is inappropriate for a general genomic feature database. Instead, we retain these kinds of data in the format in which they are originally submitted (e.g. WIG and SAM), and reference them from the database. This makes it harder to find the answer to some kinds of questions, for instance, finding the read coverage of a region across multiple submissions requires finding the SAM files for those submissions, then using tools specifically for parsing SAM data to pull out read coverage for a region, rather than writing a single query against the database. On the other hand, making these data available to the genome browser is trivial, since they are already in a supported format.
The DCC has benefitted greatly from extending Chado rather than building a new schema. Not only did we avoid the potentially huge effort of defining a new schema from scratch, we were also able to adopt existing Chado infrastructure. The ChadoXML loader gave us a portable method for passing data between components of the DCC; and we used existing tools to populate CVs in the database. We also found it easy to incorporate the publicly available Chado databases provided by WB and FB alongside our database.
In order to address support for collecting genomic features associated with short-read sequencing technologies, we would suggest that future projects investigate the development of architectures that support sharding/partitioning, making it possible to spread the load across multiple servers. We also suggest building support for querying external binary formats into the core of a data processing pipeline to enable queries of optimized formats of data that is poorly suited for a relational database. Certainly, use of Chado is recommended for any group looking to store discrete genomic annotations and when collecting ontology-based metadata, as it is optimized for both of these types of data.
Pipeline
The tracking and reporting capabilities of the pipeline have proven indispensable. The processing history is widely used, and provides feedback to data providers and DCC curators about chronic problem areas (a common case is highlighting problems in submissions that have previously been solved). Although rarely necessary, the ability to examine earlier versions of uploads is a nice feature, particularly when the original sources are unavailable. In addition, we use the timestamps to measure the speed with which submissions progress through the pipeline (Figure 4E), thereby informing our development efforts and allowing us to report pipeline performance to NIH.
Challenges of data curation and release
The automated validation and manual checklist process is nontrivial, inevitably leading to the observed lag time averaging 1 month between the first data upload and public release (Figure 4D). The lag time has decreased over the life of the project, but it has not been eliminated. It can be exacerbated by spikes in data submission, due to saturation of both the computation pipeline and curatorial resources. Additionally, data providers sometimes upload their raw and processed data files considerably in advance of the accompanying metadata, which inflates the apparent delay between upload and release. In fact, the actual time spent vetting from the first attempt at validation (implying all data has been uploaded) to public release is significantly shorter than 1 month. The mean time for all submissions is 6 h and 11 min, or after removing outliers whose processing time is >3 SDs from the mean, 1 h and 5 min (http://submit.modencode.org/submit/reports).
As modENCODE progressed, the DCC added additional requirements for data submission. The majority of these extensions were related to additional QC standards and requirements laid out by production groups or requests for enhanced reporting details on a project-wide basis. A change in QC requirements often meant that the original BIR-TAB templates were insufficient and needed modification before being acceptable for future submissions. The more rate-limiting step was the percolation of any change throughout the project. Since the personnel responsible for making the submissions were often not the scientist involved in these QC discussions, there was inevitably a communications delay to ensure everyone clearly understood the new requirements and their implementation by each affected data provider.
Though automated QC checks detected errors in data packaging and simple metadata inconsistencies, manual QC was still required. The types of errors ranged from inclusion of incorrect or duplicate files, to specifying the wrong stage or strain in the sample description. This sometimes involved going back to the data providers for clarifications to protocols or samples, or to correct errors in data files. The checklist we developed and maintained ensured that all details were correct prior to release, and consistent between submissions. Even though the manual QC process grew more involved and time-consuming as the project advanced, we believe the additional time was worth the work to turn out higher quality data for the community (approximately 1 in 20 submissions contained some type of error that was caught during this step). Having DCC curators trained in the biological techniques employed by the consortium was essential. More problematic was the handling of spikes in data submission (multiple groups depositing numerous submissions in a short time period), which challenged both our curators and computational resources. One possible solution to the computational bottleneck during high volume periods might be to temporarily deploy more computing nodes, either on the local network, or using a computing grid solution. Additionally, the more QC checks that can be automated, the less the workload on individual DCC curators.
Though we maintained a complete history of all versions of data uploaded, this is not necessarily a sustainable model for longer term projects. The difference in storage capacity needed between the minimal set of data for released submissions and the maximal set of data that includes both generated files and the full revision history of submitted data files continues to widen as the project progresses (Figure 4B). While the full data file revision history is a nice feature of our system, we believe the additional space required is not worth the cost. We would recommend developing a formal policy for removal of unused versions of data.
The role of a DCC
The modENCODE DAC was formed in Year 3 of the project, and while the DCC attempted to anticipate the needs of the DAC, running specific QC metrics was not within its initial mandate (or funding). For example, because the DCC lacked resources for signal processing, we relied on data providers to call peaks themselves. This led to peaks being generated using diverse software and options, which hindered the initial integrative analysis. Though we still require our submissions to include peak calls, we are now taking on the role of actively re-calling peaks for all submissions, in order to provide the community with consistent and comparable data.
The DCC has also taken on the role of reconfirming submission data quality. Submitting groups were responsible for conforming to modENCODE consortium-wide validation and reproducibility standards for their experimental data. Initially, the DCC did not implement checks to monitor adherence to these agreements, but after the analysis for the integrative papers, we are instituting more rigorous data quality checks. In particular, we have added the ability to record antibody QC data on the wiki, and a new validation module that checks for compliance with the data standards set by the ChIP groups. We are actively retrofitting all ChIP submissions with the relevant QC metadata. We are also implementing analysis of replicate consistency using IDR analysis (18). The general lesson is that whenever there are ‘rules’, whether for biological data or the stock market, there must also be effective monitoring in place to ensure compliance.
At the beginning of the project, we recognized the diversity in descriptive detail found in different GEO entries. Therefore, the DCC offered to submit modENCODE array-based data to GEO as a service to our data providers, and to ensure that all modENCODE data in GEO would be described uniformly. To date, we have submitted 321 submissions on behalf of the consortium. However, some data providers have not used our service, and not surprisingly we have found that the descriptions provided by these groups is incomplete, with links to the modENCODE umbrella project often lacking. We are now working on amending these GEO submissions with additional metadata.
Future work
In the time remaining, we are focusing on incorporating the results of the integrative analyses. These submissions capture correlations between the multiple different experimental approaches that have been undertaken as reported in the modENCODE scientific publications, and will inform users of the correlations that have been found (16,17).
In addition, we are in the process of migrating processed data to more permanent public repositories. The most visible community portals are WB and FB, and they are the targets for sustained archival of modENCODE’s processed data. WB has performed a shallow integration of all of our data sets into their system by mirroring our tracks on their browser. modENCODE’s updated gene models, predicted pseudogenes, non-coding RNAs and stage-specific gene expression patterns are actively being curated into WB to create a deeper long-term integration of the modENCODE data. FB is also beginning to incorporate modENCODE data; as yet, this only includes gene expression data. By the conclusion of the modENCODE project in 2012, the DCC will have migrated all data produced by the consortium to GEO and/or FB/WB for long-term accessibility. NGS data will be available through the Amazon cloud.
Conclusions
The modENCODE consortium has produced an enormous library of data to enhance the understanding of the D. melanogaster and C. elegans genomes. The diversity and complexity of data will be invaluable to the greater research community, and could only be achieved through such a large-scale project. The DCC was charged with the collection and distribution of this data catalog and subsequent genomic annotations, and we have been successful in performing this task within the context of the goals we set out to achieve.
The modENCODE DCC is a resource: a facility that is a means to an end for its users. It provides a unique link between submitters of original experimental data and its interpretation, and researchers wanting to find the results relevant to their needs. Its value is greatly enhanced in two fundamental ways: technologically, by the use of deep metadata and a CV all backed by a schema; and by the human effort of both curating the data as it streams in, and by constantly revising the technology component as the nature of the data and the queries evolves. Both the technology component and the human effort have a high cost up front, but the future payoff is very large, and can accrue over a very long time. Therefore, it is critical to make the largest, earliest possible initial releases so that the payoff period begins as soon as possible. It is natural that as more early researchers experience success with modENCODE data, the perception will filter back to the submitting groups, who will be more motivated to tailor their submissions for maximum community benefit.
Our proactive approach to collecting descriptive information seems to be successful, although only time will tell if the community will utilize the full extent of metadata collected. With careful planning, flexible methods and judicious consideration of some of the issues illustrated here, any DCC will be able to facilitate the release of large volumes of data, ultimately arming researchers with the tools to generate hypotheses and discover new scientific phenomenon.
Methods
Our software, including the Chado extension, automatic QC software, DBFields extension, liftover tool and submission pipeline, is open source and available through a public Subversion repository. Requirements and instructions for download and installation can be found on our wiki at http://wiki.modencode.org/project/index.php/Open_Source.
Controlled vocabularies and ontologies
Where possible, the DCC used existing ontologies, including the Sequence Ontology (SO) for genomic features (19), the MGED Ontology for microarray experiments (7), the Gene Ontology (GO) (20), the Ontology for Biomedical Investigations (OBI) (21) and others. Additionally, we used the lists of genes from WB/FB, strains from the worm and fly stock centers, as well as cell lines from the Drosophila Genomics Resource Center.
Data formats
Aside from raw data, there were two types of analyzed data we received: histogram plots of signal intensity (either from sequence alignment or from array probes), and the analyzed peak calls and/or genomic features. For signal intensity data, we accepted the UCSC-developed data formats BED and WIG (http://genome.ucsc.edu/goldenPath/help/wiggle.html). Many groups were already familiar with these data formats and used them for viewing their own data in the UCSC browser. For peak calls and genomic features, we accepted only the GFF3 format (http://www.sequenceontology.org/gff3.shtml). This requirement was due to our choice of Chado as a database and Gbrowse as a genome browser.
For NGS data, we accepted SAM format (22). This data format has become the standard for exchange of NGS alignments. The modENCODE DCC requires all RNA-seq alignment data to be deposited in this format, and encourages other NGS experiments to be deposited in this format as well.
We imposed some additional checks on files submitted to us in the GFF3, WIG and SAM formats. For GFF3, we require some fields to be specified that are otherwise optional. We use a ‘genome-build’ header that provides the genome build against which the GFF3 was generated, and we add special handling for a ‘parental_relationship’ attribute that specifies the type of relationship between two features linked using the existing Parent attribute. We also require that parent features appear before their child features. For WIG files, we actually relax constraints, allowing chromosome names to be specified either with or without the ‘chr’ prefix (for UCSC compatibility) and attempting to support BED-like formats labeled as WIG. For SAM files, we likewise ignore the ‘chr’ prefix on chromosome names, and require the ‘SQ’ header, which specifies the genome build.
Software used
Our use of biological software packages included the Chado database schema, the GBrowse genome browser, the samtools (22) package for SAM support and various GMOD support tools. Chado is a relational database schema developed as part of the GMOD project (13). It was chosen for the wide range of available tools, for its compatibility with the model organism databases (FB uses Chado, WB is considering migrating to it), and for the modENCODE DCC staff's familiarity with the schema. Additionally, it has very good support for CVs. We plan to submit our Chado extension to GMOD by the end of the project. Our choice of genome browser was GBrowse (23), since it is the genome browser in use at both WB and FB. It also uses strong typing of genomic features, and installation is straightforward. The samtools package provides a way to transform SAM into a more efficient binary format and supports the fast queries necessary to make the format useful as a source for GBrowse displays.
We also made heavy use of several general-purpose software packages, including the Apache web server (2.2.9), MediaWiki (1.14.0), Ruby on Rails (2.1.0) and the PostgreSQL database server (8.3). Apache is an industry standard web server, and we used additional extensions for load balancing (mod_athena) and large file uploads (mod_porter). We created a wiki with MediaWiki software for both project-wide communication, document sharing and as the repository for experimental metadata. We used the Ruby on Rails framework to build the submission pipeline interface, including much of the code for generating reports. The PostgreSQL database server was used for hosting the pipeline tracking database, GBrowse tracks and the main Chado database.
Submission statistics
All data summary statistics were based on available data in the DCC as of 31 July, 2010.
Funding
This work was supported by the National Human Genome Research Institute of the National Institutes of Health [grant number HG004269-05], Wellcome Trust [grant number 090297], and by the Director, Office of Science, Office of Basic Energy Sciences, of the U.S. Department of Energy under Contract No. DE-AC02-05CH11231. Funding for open access charge: Ontario Institute for Cancer Research; Informatics and Bio-computing; 101 College Street, Suite 800; Toronto, Ontario M5G 0A3 Canada.
Conflict of interest. None declared.
Acknowledgements
We would like to thank Seth Carbon and Erwin Frise for assistance with system administration, Kate Rosenbloom and Galt Barber for exchange of ideas with ENCODE DCC and the initial pipeline code, Ed Lee for writing a new GFF3 parser, Chris Mungall for thoughtful Chado discussions, and Ian Holmes for sharing lab space. N.L.W. designed metadata format, coordinated curator activities, curated data and drafted the manuscript. E.O.S. designed metadata format, designed and implemented software including pipeline, wiki and automated QC, and drafted the manuscript. M.D.P., Z.Z. and P.L. curated data. P.R. curated data and implemented GBrowse software. S.C. and R.S. implemented the modMine query interface and contributed to the design of the pipeline. S.M. implemented GBrowse and wiki software, and assisted with curation. R.L. and A.C. assisted with help-desk duties and QC of data. E.K. contributed to pipeline software implementation. L.S., G.M. and S.L. conceived of the study, participated in its design and coordination, and helped to draft the manuscript. All authors read and approved the final manuscript.
References
Author notes
*These authors contributed equally to this work.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}