





Abstract
Recent technological advances have lead to the ability to generate large amounts of data for model and non-model organisms. Whereas, in the past, there have been a relatively small number of central repositories that serve genomic data, an increasing number of distinct specialized data repositories and resources have been established. Here, we describe a generic approach that provides for the integration of a diverse spectrum of data resources into a unified analysis framework, Galaxy (http://usegalaxy.org). This approach allows the simplified coupling of external data resources with the data analysis tools available to Galaxy users, while leveraging the native data mining facilities of the external data resources.
Database URL:http://usegalaxy.org
Introduction
The rate of generation of genomic data is increasing at a rapid pace for both model and non-model organisms. This creates exciting opportunities for biomedical research, yet also imposes a unique set of challenges such as the need to connect biomedical scientists and their data with computational tools and to allow researchers to interactively integrate additional data from external sources into their analyses [for an excellent review see Ref. (1)]. Indeed, because the cost associated with the generation of sequence data is rapidly decreasing and because many excellent solutions exist for the managing of these data, such as InterMine (2), BioMart (3), UCSC Table Browser (4), etc, there is no surprise that specialized niche data warehouses are becoming more and more numerous.
Much of this data are readily and freely accessible to all of the general public. However, for most experimental biologists there exists a void between accessing this wealth of information and translating it into useful biological knowledge. The first problem that biologists have to cope with is the immense size of genomic data sets. These data sets often comprise entire genomes worth of information: some contain information on specific genomic elements, such as the genome wide locations of a particular human transcription factor binding site, whereas other data sets, such as multiple-species whole-genome alignments, can house information about several different organisms. Some of these data sets can easily occupy hundreds of gigabytes, causing many of these data sets, despite being freely and readily available, to go underutilized by the experimental community simply due to logistical issues related to storing massive quantities of information. Even if initial obstacles can be overcome, experimental biologists are left with few options to manipulate these data. Modern spreadsheet applications, for example, are not capable of loading a file containing all purported human polymorphisms. Another problem that is encountered is the issue of data integration and format incompatibility. Beyond simply having different types of data such as sequences, alignments and genomic intervals, there is a seemingly endless supply of data formats for each of these different datatypes. This often leads to the creation of custom one-off scripts. These small scripts are generally developed by individual labs and might only perform simple functions such as pre-parsing a file, and while these scripts may be simple, they prove to be a real hindrance to the reproducibility of research when not readily available. In cases when preprocessing scripts are available, bioinformatic tools often come with confusing or command line only interfaces. All of these interfaces are different and they are not usually designed to work together: rarely is it the case that the output of one tool can be fed directly as input into another tool. Furthermore, there are almost too many tools, making it hard for experimental biologists to know where to start or which tools are best suited for a particular analysis. These issues effectively prevent many biologists from utilizing existing genome analysis software. Thus, a unified analysis framework with a diverse set of tools capable of seamless integration with heterogeneous datasources would be highly beneficial to the biomedical research community. Here, we describe an implementation of such a solution using Galaxy (http://usegalaxy.org; 5–8).
Available both as (i) a publicly available web service (http://usegalaxy.org) providing tools for the analysis of genomic, comparative genomic and functional genomic data and (ii) a freely downloadable package (http://getgalaxy.org) that can be deployed in individual labs or on Cloud resources (9), Galaxy attempts to serve both sides of the user distribution: experimental biologists and bioinformaticians. Galaxy is not simply about accessing data and is not meant as a replacement to data warehouses as the organizations that focus on this problem are able to more effectively address the issues of storing and querying their particular data and schemas. Instead, Galaxy provides a software framework that allows the simplified coupling of external data resources with the data analysis tools available to Galaxy users, while leveraging the native data mining facilities of the external data resources. This solution is agnostic to the type of data that is returned from a particular data resource, which may itself be the result of previous analysis. By making a data resource available to Galaxy, users can simply ‘send results to Galaxy’, instead of being forced to download potentially gigabytes of data. Once data have been accessed by a user and placed into their history, it is immediately ready for analysis. Galaxy contains over a hundred analysis tools, with a concentration on providing tools that the community has established as the ‘best of’, greatly reducing the struggle to find the proper tools for a particular analysis. Galaxy is able to automatically determine data formats, and data sets can only be used as input for bioinformatic tools that are able to accept a particular format as input. In cases when the data are of the proper kind (e.g. an alignment), but the tool accepts only a particular format (e.g. a tool requires FASTA format, but the user’s data is in the MAF format), Galaxy has a collection of implicit datatype converters that handle converting the data into the format required by the tool without requiring any additional intervention by the user. Furthermore, Galaxy allows users to not only share and publish data and results (Data Libraries), but also entire analysis steps (User Histories), complete experimental protocols (Pages) and customizable plug-and-play multiple-tool analysis pipelines (Workflows).
The protocols described here allow the user to begin their analysis at either a data resource or at a Galaxy server. Separate protocols, not fully described here, are used to integrate command line analysis tools and to send data sets from Galaxy to external web applications. Furthermore, the Galaxy Upload tool, which allows data to be uploaded as a file from a user’s computer, by entering text into a form field or by providing a list of URLs, is included with the standard Galaxy distribution.
Currently, several database resources have been integrated with the public Galaxy server (http://usegalaxy.org) and are included as part of the downloadable package; a non-exhaustive list of these resources include the UCSC Table Browser (Figure 1), BioMart Central Portal, InterMine, EpiGraph (10), EuPathDB (11) and HbVar (12). Adding new data resources into Galaxy is straightforward and requires no changes to the Galaxy source code; in most cases, defining a simple XML configuration file and instructing Galaxy to load the newly defined file is sufficient to inform Galaxy of an external data resource. For occasions where a data provider is using one of the code-bases, which are already Galaxy-aware, after setting up and configuring their data resource, the process to add the new resource to an existing Galaxy instance (Galaxy instance: each occurrence of a standalone Galaxy server) requires minimal time and effort. When the data provider is hosting their resource using code which is not-yet Galaxy capable, the amount of time is dependent upon the steps required on the data provider’s part to modify and configure their own code-base; however, the time required to configure the Galaxy instance remains similar.
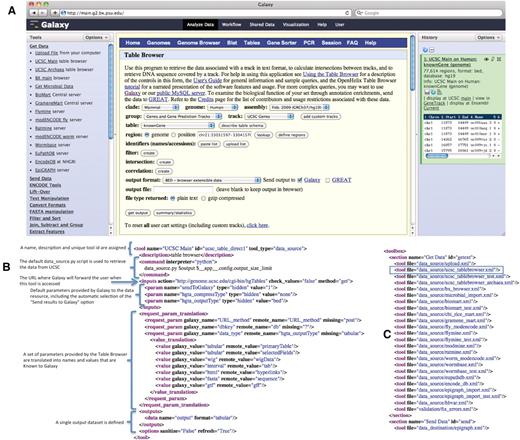
The UCSC Table Browser tool. The UCSC Table Browser tool is shown with its native interface as it appears integrated into Galaxy (A). A simplified XML configuration file (B) that describes to Galaxy how to communicate with the data resource is shown. Advanced configuration options have been used to customize data set attributes and to enhance the user experience. Values for the file format and genome build are taken from the parameters provided by the datasource and made accessible to Galaxy. Additionally, this configuration causes the ‘Send output to Galaxy’ option to be automatically selected when a user begins from within Galaxy. The addition of a single line, outlined in blue, to the tool_conf.xml file is all that is required to inform Galaxy to load the tool (C).
Methods
Depending upon data set availability, Galaxy employs two simple default protocols to communicate with external data resources: (i) synchronous and (ii) asynchronous. In the synchronous protocol, the requested data set is available from the external data resource immediately after the user has configured their desired options. When the data set is not immediately available from the external data resource, the asynchronous method is also available. While most resources utilize the synchronous approach, the asynchronous protocol has been used effectively in cases when there is significant time between the end of user interaction at the data resource and the availability of the requested data. The selection of the synchronous or asynchronous protocol is based entirely upon the technical requirements of the data resource and does not alter the user’s interaction with the data resource or Galaxy. Each individual implementation of either of these protocols is known as a datasource tool within Galaxy. Both of these methods allow the user to continue using Galaxy, while the data are being generated or transferred. Although the use of these protocols prevents the need for users to download any files onto their computer, they are able to interactively analyze their data set and can optionally download their data at any time.
User perspective
The UCSC Table Browser is an example of a tool that implements the synchronous protocol. Although this example uses the synchronous protocol, from the user’s perspective, the steps required to have data sent to Galaxy would not change if the data provider opted to use the asynchronous protocol. In this particular example, the user can begin from within the Galaxy interface (such as the one at http://usegalaxy.org) and select the UCSC Main table browser tool, or they can begin within the UCSC Table Browser interface (Figures 1A and 2). When the user begins within the UCSC Table Browser, they will need to select a checkbox, ‘Send output to Galaxy’, which informs the external resource that results should be returned to Galaxy; this checkbox is located on the first page of the Table Browser interface and is automatically selected by default when a user begins within Galaxy. After ensuring that the option to have results sent to Galaxy is selected, the user is able to customize the parameters of their query within the native Table Browser interface in the same manner as if Galaxy was not a factor. When the user is ready to retrieve their data from the Table Browser, they use the ‘get output’ button, which presents an additional page that allows the selection of additional data formatting options.
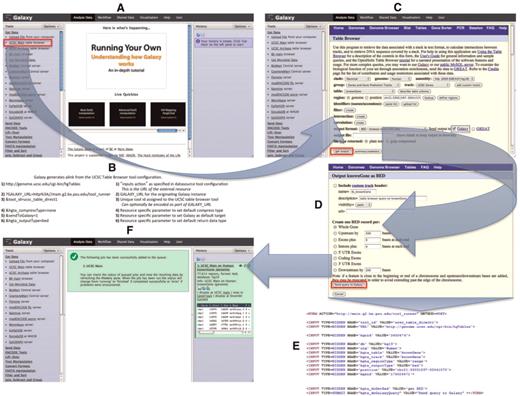
UCSC Table Browser as a synchronous data resource example. An overview of a typical synchronous data resource tool, with the UCSC Table Browser as an example, is shown here. Based upon the XML configuration file for the UCSC Table Browser tool (Figure 1), Galaxy creates a new tool as a link (outlined in red) that references the data resource under the Get Data tool section (A). An example of the link (B) that is generated is described along with the parameters of which it is composed; several of the parameters provided in the tool XML configuration customize the initial interface of the external resource. By accessing the link, the user is forwarded within their web-browser to the native UCSC Table Browser interface (C). Once the user is satisfied with their query configuration and has selected the desired formatting options (D), the UCSC Table Browser generates a form (E; for brevity, some parameters have been removed from the original HTML) with an action that points to the Galaxy server. When Galaxy receives the post (F), a new data set is created in the user's history. Galaxy collects the parameters provided within the request and executes a process in the background that resubmits these parameters back to the Table Browser at the location specified by the provided URL parameter; the response from the Table Browser is the content that Galaxy will use to populate the new data set.
At this point, there is a minor deviation from the standard user experience within the Table Browser interface, where ordinarily there would be a button that allows the user to download the data set onto their computer (e.g. ‘get BED’), there is instead a button labeled ‘Send query to Galaxy’ (Figure 2D) that will direct the user along with their data set to the Galaxy server. The user is now able to interactively analyze their data set without needing to download copies onto their computer, but they are able to download their original data, intermediate analysis steps or final results at any time.
Although the UCSC Table Browser requires the user to declare that they want the results of their query to be returned to Galaxy before selecting the final formatting options, this need not be the case. For example, Intermine servers, which support returning data sets to Galaxy, place the option on the results page within the ‘Export’ menu. Galaxy places no demands on the particulars of the user interface and we think that the developers of each data resource are most able to make these decisions based upon their individual requirements and specifications.
Synchronous data resources
The synchronous data connection protocol should be used for circumstances when user requested data is available in real-time. This overview of the protocol commences with the case where a user starts at a Galaxy server, but is applicable for when a user starts from the external database resource, in which case default parameter values are used to indicate the target Galaxy instance. Figure 2 provides an overview of this process using the UCSC Table Browser tool as an example. To begin, a user selects the datasource tool from Galaxy’s tool menu, found in the left pane of the web interface. This causes Galaxy to send the user to the external data resource’s URL (specified as the ‘inputs action’ attribute in the individual datasource tool configuration XML file) along with the parameter GALAXY_URL in this request. The value of the GALAXY_URL parameter contains the URL where Galaxy will expect a response to be sent at some later time. It is the external site’s responsibility to keep track of this URL as long as the user navigates the external resource. When a user begins from the external data resource, a default GALAXY_URL as defined by the external resource, is used and typically references the main public Galaxy server. It is the reliance on this configurable parameter that allows many Galaxy instances located at different URLs around the world to interact with the same data resource without requiring a centralized Galaxy server.
As the user navigates the external data resource, it behaves exactly as it would if the request had not originated from Galaxy. At the point where parameter submission would ordinarily return data to the user, the external data resource will have to instead post these parameters to the URL that was sent in the GALAXY_URL parameter, additionally providing a parameter URL that contains the location from which to request the data from the external resource. Typically, this would require that the action attribute of the form that normally generates data to be pointed to the value provided by the GALAXY_URL parameter with the usual, non-Galaxy interactive, form action target sent as the URL parameter. By relying on a form or link, which the user accesses within their own web-browser, locally installed Galaxy instances are able to connect with external data resources without requiring additional firewall configuration. For example, by default, a freshly installed personal Galaxy instance binds to localhost on port 8080, which is the basis for the GALAXY_URL parameter. When the user submits the form, the request occurs from within their own browser. As long as the user is able to access the remote host from their current network connection, then they are able to load data sets into their local Galaxy instance from that external resource.
When Galaxy receives the parameters, it will run a URL retrieval process in the background that will submit the parameters to the external resource, located at the value of the provided URL parameter. The response from the external data resource should contain the desired data content that Galaxy will save in the user’s current workspace (known as a user’s History). For production servers, this background process can be dispatched to compute nodes which have network access to the external resource; these compute nodes do not need public IP addresses and can make full use of e.g. network address translation (NAT), as the data connection is initiated by the node.
Asynchronous data resources
The asynchronous protocol should be used when the user requested data is not available in real-time, because, for example, the external datasource needs to execute a background process to generate the data. This process operates similarly to the synchronous protocol, with the exception being that the external resource will have to later notify Galaxy with the location of the data.
The same steps are followed as in the synchronous data protocol, but, instead of the user requested data being available at the URL parameter provided by the external resource, a different series of communication events occur. In lieu of the final step in the synchronous protocol, Galaxy will create a new GALAXY_URL parameter that will uniquely identify the target data set to be populated with the not-yet-generated data and will send this information and the user-specified parameters back to the external resource located at the provided URL parameter (in the synchronous protocol, this URL would contain the data used to populate the Galaxy data set). This should cause the external data resource to execute the background processes required to generate the data content. At this point, Galaxy has created a data set object to store the data content and is waiting for notification from the external resource that the data is ready. This approach prevents the need for Galaxy to continuously poll the resource. Inter-process communication is performed via very simple text outputs. Commands that have been executed correctly may write any kind of text messages; if the text ends with the word OK, it will be considered a successful submission. Messages that do not end with OK will be treated as errors. There is no requirement on interpreting any of the messages as they primarily serve informational and debugging purposes.
When the data generated by the external resource is ready, the resource will have to connect to the URL specified in the most recently provided GALAXY_URL and provide STATUS and URL parameters. Galaxy will then make a background request to fetch the data stored at the location specified in URL. Both parameters STATUS and URL must be present. If STATUS is different than OK, then the user’s data set will be marked as failed and data will not be retrieved. In the case of an error, the external data resource may include a more detailed value for STATUS, since this value will be stored and displayed to the user as the reason for the failure.
Advanced data resource configuration
While the steps described so far are entirely sufficient for getting data content into Galaxy, often additional information about the data is desired for maximum usability, such as data format, source genome build, data set name and additional free-form information. Galaxy is able to parse the parameters provided by the external resource for this information. By default, Galaxy will use the values provided in the data_type, dbkey, name and info parameters, respectively, for this purpose (Table 1). The external resource does not need to use these exact parameter names, as the datasource tool configuration file can provide parameter name and value translations that can be used for this purpose. Name translations function to provide a parameter differently named by the external resource as one of the parameters with special function. Value translations provide a different value to Galaxy for a particular parameter than what was provided by the external data resource; e.g. this can be useful for mapping between non-standard genome build aliases.
Data resources can provide parameters to customize how data sets are added to a user's History
| Parameter name | Usage |
|---|---|
| Name | The external resource can provide a descriptive name for the retrieved data set. If not provided, a default name based upon the name provided in the XML tool configuration is used. |
| Info | A free-form text string that a resource can use to provide additional information about the data set. |
| data_type | The type of data returned to Galaxy. Examples include bed, sam, gff and maf. |
| Dbkey | If the data belongs to a single reference genome, this string is used to store this information. Examples include hg18, mm9 and canFam2. |
| URL | The user’s history will be populated with a new data set containing the results returned by submitting all provided parameters to this URL. |
| Parameter name | Usage |
|---|---|
| Name | The external resource can provide a descriptive name for the retrieved data set. If not provided, a default name based upon the name provided in the XML tool configuration is used. |
| Info | A free-form text string that a resource can use to provide additional information about the data set. |
| data_type | The type of data returned to Galaxy. Examples include bed, sam, gff and maf. |
| Dbkey | If the data belongs to a single reference genome, this string is used to store this information. Examples include hg18, mm9 and canFam2. |
| URL | The user’s history will be populated with a new data set containing the results returned by submitting all provided parameters to this URL. |
Data resources can provide parameters to customize how data sets are added to a user's History
| Parameter name | Usage |
|---|---|
| Name | The external resource can provide a descriptive name for the retrieved data set. If not provided, a default name based upon the name provided in the XML tool configuration is used. |
| Info | A free-form text string that a resource can use to provide additional information about the data set. |
| data_type | The type of data returned to Galaxy. Examples include bed, sam, gff and maf. |
| Dbkey | If the data belongs to a single reference genome, this string is used to store this information. Examples include hg18, mm9 and canFam2. |
| URL | The user’s history will be populated with a new data set containing the results returned by submitting all provided parameters to this URL. |
| Parameter name | Usage |
|---|---|
| Name | The external resource can provide a descriptive name for the retrieved data set. If not provided, a default name based upon the name provided in the XML tool configuration is used. |
| Info | A free-form text string that a resource can use to provide additional information about the data set. |
| data_type | The type of data returned to Galaxy. Examples include bed, sam, gff and maf. |
| Dbkey | If the data belongs to a single reference genome, this string is used to store this information. Examples include hg18, mm9 and canFam2. |
| URL | The user’s history will be populated with a new data set containing the results returned by submitting all provided parameters to this URL. |
At this time, it is worth discussing the discrepancies in data between various providers. Among these differences are not only the previously mentioned genome build identifiers (dbkey), but also chromosome names and coordinate systems. Although differences between coordinate systems can be resolved by adhering to standard formats (e.g. BED, GTF, SAM/BAM, MAF, etc.), properly handling the other differences is not as straightforward. An example of the discrepancy found in genome builds is seen with the latest human reference, which may be referred to as GRCh37, hg19 or others. Likewise, chromosomes could be referred to as e.g. chr1, chrX, chrM, etc. or as 1, x, MT, etc. These issues pose significant challenges when attempting to work with data sets from providers that use different nomenclature systems. While Galaxy can provide tools to allow users to manually modify data sets and even automatically (see next paragraph) modify data contents, we think that it is in the best interest of the community to adopt a set of standard naming rules. Even without considering Galaxy, the community would be well served if data providers would agree to use the same naming conventions.
By default, a standard Galaxy command line executable (data_source.py) is run in the background to fetch the data content from the external data resources. This executable can be replaced on a per resource basis, with any program or script of the implementer’s desire. This can be particularly useful when the data provided by the external resource needs to be transformed in some fashion before being acceptable for downstream analysis and requiring the user to execute a separate formatting tool is not desired. A custom executable can also be used in cases when the external resource initially provides e.g. an XML file that describes the location of several files.
Additional approaches
It is our experience that by providing these two simple protocols, the majority of external data resources can be seamlessly integrated into Galaxy. However, integrating external data resources into Galaxy is not limited to the two standard protocols described here.
In cases when the aforementioned protocols are not adequate for a particular external data resource, the resource can be integrated as a standard Galaxy tool. While exhaustively defining a generic Galaxy tool is outside of the scope of this manuscript, briefly, a Galaxy tool is composed of any command line accessible executable and a tool configuration file that describes the input parameters and output data sets to be created; see Figure 3 for an example of a simple EFetch-based NCBI (13) sequence retrieval tool which does not utilize the standard Galaxy protocols. Additionally, Galaxy provides data library functionality that presents pre-cached access to static data sets, which may only be originally available as files from the data provider. Figure 4 shows an example of this, where the 1000 Genomes project pilot data (14) was loaded directly into Galaxy from an FTP server. The use of a Galaxy data library has the added effect of preventing duplication of data sets on disk when a user imports a data set into a history. Although each copy of a particular imported data set shares a reference to the same file on disk, the user is free to modify the metadata and attributes of their copy as they see fit.
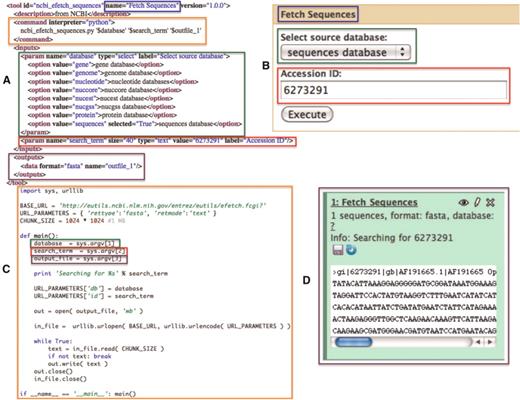
A simple NCBI sequence retrieval tool. This minimal tool interface (A: Galaxy tool description and B: Galaxy generated user interface) consists of a single textbox that allows the user to manually enter an accession number and a select list that allows the user to specify the target sequence database to search. When a user executes this tool, a simple script (C) is run by Galaxy which fetches the FASTA sequence data (D) for the user provided accession number. Color-matched boxes have been added to indicate the interrelatedness of various elements of the panels.
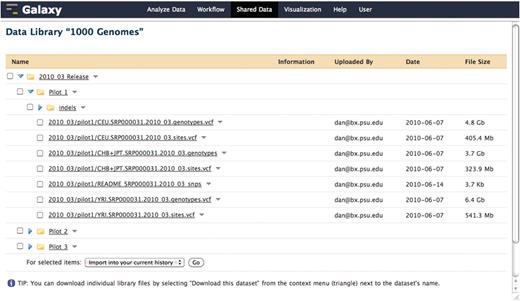
A Galaxy library containing pilot data from the 1000 Genomes project. This data was loaded directly into a Galaxy data library from the 1000 Genomes project FTP server. When a user imports a data set from a library, the underlying file on disk is not copied. Although each copy of a particular imported data set shares a reference to the same file on disk, the user is free to modify the metadata and attributes of their copy as they see fit.
Conclusion
Galaxy is a powerful platform that provides biomedical researchers with integrated access to data resources, a best-practices collection of analysis tools and visualization resources. While unsupervised integration has been shown to be an effective initial analysis step, it is supervised integration that affords the greatest advance to biological understanding (1). Using Galaxy, researchers are able to directly query data providers using the native data mining facilities provided by the external resource. By relying on the external resources to provide the querying interface, the time required to configure Galaxy to communicate with an external resource is minimized while simultaneously imparting full control of data access to the resource curators; for example, changes made to the external resource interface are reflected instantly without additional effort required in the Galaxy instance.
We have presented two standardized protocols that simplify the addition of external data providers into Galaxy: synchronous and asynchronous. These protocols broadly handle the two general cases of data set availability: real-time or delayed, respectively. Facilities are provided to allow the external resource to specify metadata of the requested data such as format and reference genome. Although the two protocols are able to cover the majority of external data providers, we have briefly presented alternatives. In cases when the external data sets are available only as files, a Galaxy data library can be used; the use of a Galaxy library has the added benefit of preventing the duplication of primary data set file content on the Galaxy server. An example of using a standard Galaxy tool to access remote resources was also briefly presented as a means to query external resources that are not Galaxy-aware. In addition to this manuscript, a step-by-step example filled tutorial, titled DataSources, is available from the Galaxy wiki (available at http://getgalaxy.org).
Acknowledgements
Efforts of the Galaxy Team (Enis Afgan, Guru Ananda, Dannon Baker, Dan Blankenberg, Ramkrishna Chakrabarty, Dave Clements, Nate Coraor, Jeremy Goecks, Jennifer Jackson, Sergei Kosakovsky Pond, Greg Von Kuster, Ross Lazarus, Kanwei Li, Anton Nekrutenko, James Taylor and Kelly Vincent) were instrumental for making this work happen.
Funding
The Beckman Foundation Young Investigator Award (to A.N.); National Science Foundation (DBI 0543285) and National Institutes of Health (HG004909 to A.N. and J.T.); National Institutes of Health (HG005133 and HG005542 to J.T. and A.N.); the Penn State University and the Huck Institutes for the Life Sciences (to A.N.); the Emory University (to J.T.). Additional funding is provided, in part, under a grant with the Pennsylvania Department of Health using Tobacco Settlement Funds. The Department specifically disclaims responsibility for any analyses, interpretations or conclusions. Funding for open access charge: Penn State University.
Conflict of interest. None declared.

{kind=link}
{kind=link}
{kind=link}
{kind=link}