





Abstract
The Comprehensive Phytopathogen Genomics Resource (CPGR) provides a web-based portal for plant pathologists and diagnosticians to view the genome and trancriptome sequence status of 806 bacterial, fungal, oomycete, nematode, viral and viroid plant pathogens. Tools are available to search and analyze annotated genome sequences of 74 bacterial, fungal and oomycete pathogens. Oomycete and fungal genomes are obtained directly from GenBank, whereas bacterial genome sequences are downloaded from the A Systematic Annotation Package (ASAP) database that provides curation of genomes using comparative approaches. Curated lists of bacterial genes relevant to pathogenicity and avirulence are also provided. The Plant Pathogen Transcript Assemblies Database provides annotated assemblies of the transcribed regions of 82 eukaryotic genomes from publicly available single pass Expressed Sequence Tags. Data-mining tools are provided along with tools to create candidate diagnostic markers, an emerging use for genomic sequence data in plant pathology. The Plant Pathogen Ribosomal DNA (rDNA) database is a resource for pathogens that lack genome or transcriptome data sets and contains 131 755 rDNA sequences from GenBank for 17 613 species identified as plant pathogens and related genera.
Database URL:http://cpgr.plantbiology.msu.edu.
Introduction
The genomes of plant pathogens range from small nucleic acid molecules in viruses and viroids, typically in the kilobase (kb) size range or smaller, to large, complex genomes in the megabase (Mb) range in higher eukaryotic pathogens including fungi, oomycetes and nematodes. Due to their smaller size, genome sequences for viruses and viroids can be determined using conventional molecular biology and first-generation sequencing methods such as Sanger sequencing. The genomics era for cellular plant pathogens began in 1999 with the publication of Expressed Sequence Tags (ESTs) from Phytophthora infestans (1), which are single pass sequences of cDNAs that are highly informative with respect to gene discovery and assessment of expression levels. An EST collection typically contains redundant, as well as partial sequences of the same transcript (mRNA) concomitant with sequencing errors due to the single pass nature of the method. The issues of redundancy, quality and length can be addressed through the clustering and assembly of the primary ESTs into longer, more accurate consensus sequences that are termed unigenes, transcript assemblies or tentative consensus sequences (2–4). EST approaches for gene discovery are well established in phytopathological research; these approaches have also yielded new information on pathogenesis (5–9).
The first cellular plant pathogen genome sequence obtained was Xylella fastidiosa (10) in 2000. The X. fastidiosa is a xylem-limited, insect-vectored bacterium and causal agent of citrus variegated chlorosis and Pierce's disease of grape. In the 11 years, since the X. fastidiosa genome was published, genome sequences have been reported for a diverse range of plant pathogens including bacterial, fungal, oomycete and nematode pathogens. Access to the genome sequence, and the associated annotation of genes and regulatory features, has provided major insight into the mechanisms of virulence and pathogenicity, mediators of host-specificity, pathogen biochemistry and physiology and adaptation to ecological niches (11–17). Genome-sequencing efforts have primarily involved culturable organisms because high-purity DNA preparations from a single isolate can be obtained. With the advent of improved sequencing technologies, coupled with increased throughput and decreased costs (18–20), metagenomics, in which a population of organisms is sequenced en masse, can be employed. The genome sequence of Candidatus Liberibacter asiaticus was obtained using metagenomics by whole-genome shotgun sequencing of an infected psyllid (21), thereby bypassing the need for culturing of the pathogen. Analysis of the Ca. L. asiaticus genome revealed a highly reduced genome that is consistent with its lifestyle as an intracellular pathogen and insect symbiont. Intriguingly, analysis of the gene complement involving biochemical processes revealed auxotrophy for five amino acids that could improve efforts to culture this organism.
Whereas analysis of a single genome will yield its gene complement and insight into biological processes, comparative analyses of two or more genomes permits identification of conserved and divergent features. Comparative genomics can be used to investigate similarities and differences among species, genera and higher order clades or to reveal intraspecific variability. For example, analysis of the genomes of two Xanthomonas isolates, Xanthomonas campestris pv. campestris and Xanthomonas axonopodis pv. citri, the causal agents of citrus canker and black rot of crucifers, respectively, revealed not only large sets of orthologous genes, but also isolate-specific genes (22). With further expansion of genome sequencing, the ‘core’ genome, that represents genes required and highly conserved in all isolates of a species, and the pan-genome, which includes the core genome, as well as all genes that vary among isolates, can be refined as demonstrated for human microbial pathogens (23). Comparative analyses can also be used to refine and standardize annotations across related genomes (24–26).
Several databases for plant pathogen genomes have been developed. These are primarily species- or clade-oriented and were developed as part of an initial-associated genome-sequencing project such as the VBI Microbial Database (VMD) (27) and the Fusarium graminearum Genome Database (FGDB) (28, 29). Plant pathogen genomes are also included in databases that encompass a wider range of taxa such as the e-Fungi database (30), the Fungal Genome Initiative at the Broad Institute (http://www.broadinstitute.org/scientific-community/science/projects/fungal-genome-initiative/fungal-genome-initiative) and the Fungal Genomic Program at the Joint Genome Institute (JGI; http://genome.jgi-psf.org/programs/fungi/index.jsf) that house fungal genomes, as well as the Integrated Microbial Genomes (IMG) (31), which supports a wide range of microbial genomes. Other databases are available that specialize in particular types of data, such as pathogen–host interactions (PHI-base) (32), pathogenic fungal ESTs (COGEME) (33), fungal secretome (FSD) (34) and a plant pathogen expression profile database (PLEXdb) (35). These specialized databases tend to store only particular types of data dedicated to specific phytopathogens. Even though some of these databases contain multiple genomes, the distribution of genomic data and lack of a comparative analytical framework makes systematic comparisons of genomes challenging.
To address these limitations, we developed the Comprehensive Phytopathogen Genome Resource (CPGR), which provides a central portal to information, as well as data for plant pathogen genomes. The CPGR Genome Warehouse provides links to all viroid, viral, bacterial, oomycete, fungal and nematode plant pathogens genome and/or transcriptome projects. Genome sequence data from publicly available bacterial, fungal and oomycete projects are accessible on the CPGR through a suite of search, analysis and display tools with nematode genome sequences to be released in the future. To provide higher quality consensus sequences for EST-based projects, we have constructed the Plant Pathogen Transcript Assemblies database with annotated sequences for eukaryotic plant pathogens. With the growing importance of genomic sequence data in applied plant pathology research, the CPGR provides data sets including the rDNA database for plant pathogens, the simple sequence repeat (SSR) or microsatellite tool and a list of unique loci for complete bacterial genomes to facilitate development of genomic-based diagnostic markers.
Materials and methods
CPGR data sources and database design
The data source for the CPGR is composed of four separate databases: Annotation, Plant Pathogen Transcript Assemblies, rDNA and the Genome Warehouse. The Plant Pathogen Transcript Assemblies, rDNA database and Genome Warehouse use three separate MySQL relational databases with custom schemas, whereas the Annotation database uses the Chado schema made available by the Generic Model Organism Database (GMOD) Project that develops and deploys open-source software for genome sequence, annotation and analysis projects (36).
CPGR Warehouse
The CPGR Warehouse is a database that stores information on plant pathogen genome and EST sequencing projects and is updated at least twice a year. For other taxa, information on new genome projects and updates for existing projects is collected from several web sources such as NCBI Genomes (http://www.ncbi.nlm.nih.gov/sites/genome) and Genomes Online Database (GOLD; http://www.genomesonline.org/), as well as direct user submissions. New and updated data are converted into a custom XML format and kept under version control to permit tracking of modifications. A custom Perl script is used to load the XML files into the Warehouse MySQL database that serves as the source for the Warehouse web page data.
Annotation database
The Annotation database contains genome annotation data and is updated using a custom Perl pipeline that first downloads new and updated annotation data sets from A Systematic Annotation Package database (ASAP, 37) and GenBank and then loads the annotation to the CPGR database. In addition to sequence information, ASAP is comprised of a MySQL database containing features and associated annotations. The feature and annotation types include all those available in GenBank, as well as custom features and annotations for added value. With an evidence-based model of annotation, links to PubMed articles and other sources are provided for every line of annotation, providing the user with a means of judging their validity in addition to allowing rapid access to further information about the feature. Annotations also contain an identifier of the species from which it was originally described to allow the user to assess the appropriateness of the annotation to the feature in question. Annotations can be added to the database by registered users and are curated by members of the ASAP staff for clarity and applicability. Annotated genomes in the ASAP database are then imported into the CPGR using a custom Perl pipeline. First, the genomes are exported from ASAP in GenBank format and converted to GFF3 format. The GFF3 is then imported into two CPGR databases. One database is a Bio::DB::SeqFeatureStore used as the backend for the Generic Genome Browser. The other database uses the GMOD Chado schema and is used as the main annotation database for the CPGR analysis and web display. The transcript and protein sequences are then dumped for each locus from the Chado database in FASTA format. A range of protein-level analyses are then run and loaded into the Chado database including: UniRef100 BLASTP, InterPro, PFAM, TMHMM and All-vs-All BLASTP search. The end user can access the annotation database through the CPGR website.
Transcript assemblies
For the Plant Pathogen Transcript Assemblies, ESTs and mRNAs are downloaded from GenBank and cleaned of low quality and contaminating sequences using Seqclean (http://compbio.dfci.harvard.edu/tgi/software/). Transcript assemblies (TA) are constructed using the TIGR Gene Indices Clustering (TGICL) pipeline (3) with a minimum sequence overlap identity between two sequences of 95%, a minimum overlap between sequences of 50 bp and a maximum overhang of non-overlapping sequences of 20 bp. TAs and remaining singleton ESTs are functionally annotated by searching against UniRef100 (38) using BLASTX with an E-value cutoff of 1 × 10−5. Low complexity sequences are masked in the UniRef100 database using the program Seg (39). SSRs are identified using a custom Perl script using minimum repeat sizes of 10 mononucleotides, 5 dinucleotides and 4 tri-, tetra-, penta- and hexanucleotides. Primers flanking the SSRs are picked using Primer3 (40). The TAs and annotation data are stored in the MySQL database using a custom schema developed for the Plant Transcript Assemblies project (2) which clusters and assembles ESTs into nonredundant consensus sequences.
Ribosomal DNA database
The ribosomal DNA (rDNA) database is composed of a MySQL database and a web-based tool for querying the database. A central component of the rDNA database is a table containing the names and NCBI taxon identifiers for plant pathogens. Plant pathogen names were curated from master lists obtained from the National Plant Diagnostic Network (http://www.npdn.org) and the American Phytopathological Society (http://www.apsnet.org/publications/commonnames/Pages/default.aspx) with additional entries and corrections submitted by users. Another table in the rDNA database links the pathogens to plant diseases. The rDNA database is updated on a regular basis with new rDNA sequences downloaded from GenBank and loaded into the database using a custom Perl script.
Genome annotation
Functional annotation for bacterial genes was obtained from the initial GenBank import or through curation in the ASAP database. Annotations can be transferred between orthologous features within the ASAP database to standardize them between genomes. Orthologs are initially determined as best reciprocal BLAST hits and are confirmed through manual examination or other processes which take feature synteny into consideration, such as whole-genome Mauve alignments (41). This transfer of annotations can take place for features across an entire genome, or between individual features based on a curator's discretion, and may include the full set of annotations or a specific subset. Fungal and oomycete genomes were downloaded from GenBank and converted to GFF3 using GMOD tools. The annotation was then loaded into the annotation database using the pipeline described above. Development of pipeline to annotate publicly available eukaryotic plant pathogen genome assemblies that do not have annotation available in GenBank is currently in progress.
Identification of orthologous groups for bacterial genomes
Bacterial genomes were grouped based on taxonomic class (Alphaproteobacteria, Betaproteobacteria, Gammaproteobacteria, Actinobacteria, Mollicutes) and orthologous/paralogous clusters generated using the OrthoMCL algorithm using the default parameters (42, 43). Orthologs and paralogs as defined by the OrthoMCL program were parsed and stored in an SQLite database using a custom schema.
Annotation of pathogen virulence factors in bacterial genomes
Putative virulence factors are identified through a manual examination of literature, typically as features that, when disrupted, result in a decrease or absence of disease symptoms in the host plant. These features receive a specific annotation that permits rapid querying within a particular genome for every feature that contains it. A list of these features is provided at the CPGR website (http://cpgr.plantbiology.msu.edu/cpgr_asap_vf.shtml) with links to feature pages on both the CPGR and ASAP sites.
Search tools
A suite of tools is available that allows users to search the loci based on sequence and functional annotation. BLAST searches are supported using WU-BLAST (44). Annotation databases storing the bacterial, fungal and oomycete annotation can be searched via locus identifiers, putative function, Pfam domain, Interpro domain and orthologous group membership (bacteria only). Upon submission of the input, the underlying database is searched and the results are parsed and displayed in the results page.
The SSR Candidate Marker Search Tool (http://cpgr.plantbiology.msu.edu/cpgr_ssr_marker_pred.shtml) is a web-based tool that allows the user to scan a submitted sequence for SSRs and automatically design primers flanking the SSR using Primer3. The tool allows the user to select the minimum number of repeats for monomer through hexamer SSRs, the Primer3 parameters and the sequences to be searched in FASTA format. A maximum of 50 sequences can be submitted with a total file size of 1 MB. Upon submission of the sequences, the tool scans for SSRs using a custom Perl script and generates a temporary Primer3 input file. Primer3 is then run on the temporary file and the output is parsed and displayed in the results page.
As the number of bacterial species with genome sequence is extensive, we have developed the Unique Loci List Query Page (http://cpgr.plantbiology.msu.edu/cgi-bin/bact_gateway.pl?page=unique_loci) that displays lists of unique loci for each bacterial genome in the CPGR Warehouse. The unique loci are identified by performing an All-vs-All BLASTP (E-value cutoff of 1 × 10−5) search of the protein sequences of the entire set of bacterial loci populated within the Annotation database. A custom Perl script is used to parse the BLASTP output, identify loci with self-hits and load the information to a dedicated database table.
Results
CPGR Warehouse
We have created a ‘Genome Warehouse’ that is a web-based information portal for plant pathogens in which genome or large-scale transcriptome sequence data sets for viroids, viruses, oomycetes, nematodes, fungi and bacteria are collated to provide the community with the status, source and metadata regarding each genome project. By querying public databases such as NCBI and GOLD, we identified 778 genome or transcriptome sequence data sets, as well as 28 genome or transcriptome projects in progress (Table 1). Outside of ‘in progress’ genome/transcriptome projects, the genome sequence data sets can be in either a ‘finished’ state in which the underlying sequence quality is high or in a ‘draft’ state in which the genome sequence has been generated through a whole-genome shotgun sequencing approach and gaps and errors such as base calls and misassemblies are known to be present. Transcriptome projects (for more details see below) are collated in the Warehouse as EST projects.
List of genomes and transcriptome projects within the Comprehensive Phytopathogen Genome Resource Warehouse
| Taxonomic group | Status | |||
|---|---|---|---|---|
| Finished | Draft | In progress | ESTs | |
| Bacteria | 34 | 15 | 14 | 0 |
| Fungi | 7 | 13 | 14 | 22 |
| Nematodes | 0 | 2 | 0 | 14 |
| Oomycetes | 0 | 6 | 0 | 6 |
| Virus | 623 | 0 | 0 | 0 |
| Viroid | 36 | 0 | 0 | 0 |
| Total | 700 | 36 | 28 | 42 |
| Taxonomic group | Status | |||
|---|---|---|---|---|
| Finished | Draft | In progress | ESTs | |
| Bacteria | 34 | 15 | 14 | 0 |
| Fungi | 7 | 13 | 14 | 22 |
| Nematodes | 0 | 2 | 0 | 14 |
| Oomycetes | 0 | 6 | 0 | 6 |
| Virus | 623 | 0 | 0 | 0 |
| Viroid | 36 | 0 | 0 | 0 |
| Total | 700 | 36 | 28 | 42 |
List of genomes and transcriptome projects within the Comprehensive Phytopathogen Genome Resource Warehouse
| Taxonomic group | Status | |||
|---|---|---|---|---|
| Finished | Draft | In progress | ESTs | |
| Bacteria | 34 | 15 | 14 | 0 |
| Fungi | 7 | 13 | 14 | 22 |
| Nematodes | 0 | 2 | 0 | 14 |
| Oomycetes | 0 | 6 | 0 | 6 |
| Virus | 623 | 0 | 0 | 0 |
| Viroid | 36 | 0 | 0 | 0 |
| Total | 700 | 36 | 28 | 42 |
| Taxonomic group | Status | |||
|---|---|---|---|---|
| Finished | Draft | In progress | ESTs | |
| Bacteria | 34 | 15 | 14 | 0 |
| Fungi | 7 | 13 | 14 | 22 |
| Nematodes | 0 | 2 | 0 | 14 |
| Oomycetes | 0 | 6 | 0 | 6 |
| Virus | 623 | 0 | 0 | 0 |
| Viroid | 36 | 0 | 0 | 0 |
| Total | 700 | 36 | 28 | 42 |
For each entry in the CPGR Warehouse, we provide the name of the organism, NCBI Taxonomy identifier (http://www.ncbi.nlm.nih.gov/taxonomy), the group of organism (Virus, Viroid, Bacterium, Fungus, Oomycete, Nematode), disease caused by the organism, status of the project (Finished, Draft, In progress, EST), genome size or number of ESTs, GenBank accession numbers (if available), PubMed accession number (if available) and Genome Center or Laboratory that performed the work (Figure 1). When available, this metadata is hyperlinked to appropriate web-based links for additional information for the user. A tool is provided at the top of the CPGR Warehouse page to allow filtering of the Warehouse contents based on taxonomic group or status. Each column in the Warehouse display page can be sorted.
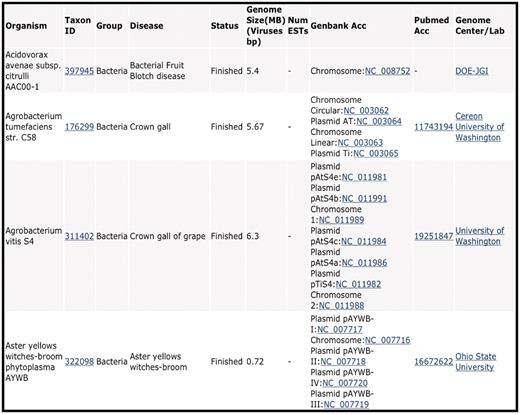
CPGR Warehouse. Output derived from filtering the CPGR Warehouse for bacterial genome projects. The output has been alphabetized based on the organism name and only the first four genome projects are listed. The organism name including strain designation, NCBI Taxon ID (http://www.ncbi.nlm.nih.gov/taxonomy), warehouse group, disease, genome status, genome size, number of ESTs (none in this example as these are genome projects), GenBank accession numbers hyperlinked to GenBank http://www.ncbi.nlm.nih.gov/genbank/), Pubmed accession numbers hyperlinked to Pubmed (http://www.ncbi.nlm.nih.gov/pubmed) and the Genome Center or laboratory that completed the work are provided in the output.
TAs
Complete genome sequences are not available for all organisms, yet large-scale sequence data sets exist in the form of mRNAs and ESTs that sample the genic regions of the genome. Within the CPGR, all publicly available mRNAs and ESTs from eukaryotic plant pathogens are downloaded from GenBank and the NCBI dbEST database (http://www.ncbi.nlm.nih.gov/projects/dbEST/), respectively. The data are clustered and assembled into a set of assemblies and singleton ESTs using the TGICL package (3). The resulting TAs (or contigs), along with the unassembled singleton ESTs, are annotated for function through searches against a protein database and loaded into the Plant Pathogen Transcript Assemblies database. Each TA is numbered uniquely (e.g. TA279_62688) (Figure 2) in which 279 represents a unique identifier within a specific TA build and the 62688 is the NCBI taxon identifier for the species (e.g. Blumeria graminis f. sp. hordei from NCBI Taxonomy). Singleton ESTs are represented through their GenBank accession numbers, e.g. EB530721. The current TA database contains transcripts from 82 different species (Table 2) representing 811 Mb of total sequence.
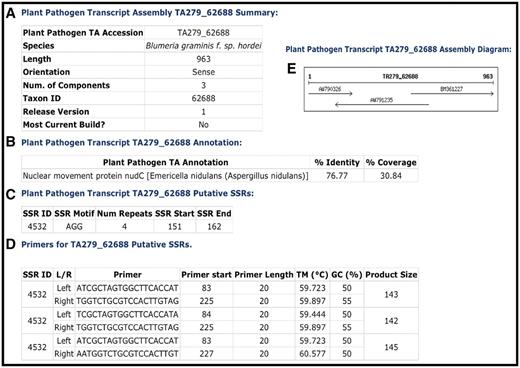
Example of a CPGR plant pathogen transcript assembly. (A) Summary metrics on Blumeria graminis f. sp. hordei TA279_62688. (B) Annotation of TA279_62688. (C) Putative simple sequence repeats for TA279_62688. (D) Primers predicted with Primer3 for putative SSR 4532. (E) Assembly diagram for TA279_62688.
Transcript assemblies of phytopathogens
| Group | No. of species | No. of ESTs and mRNAs | No. of Transcript Assemblies |
|---|---|---|---|
| Fungi | 58 | 1 049 338 | 401 952 |
| Nematodes | 17 | 162 300 | 76 681 |
| Oomycetes | 7 | 317 936 | 126 336 |
| Total | 82 | 1 529 574 | 604 969 |
| Group | No. of species | No. of ESTs and mRNAs | No. of Transcript Assemblies |
|---|---|---|---|
| Fungi | 58 | 1 049 338 | 401 952 |
| Nematodes | 17 | 162 300 | 76 681 |
| Oomycetes | 7 | 317 936 | 126 336 |
| Total | 82 | 1 529 574 | 604 969 |
Transcript assemblies of phytopathogens
| Group | No. of species | No. of ESTs and mRNAs | No. of Transcript Assemblies |
|---|---|---|---|
| Fungi | 58 | 1 049 338 | 401 952 |
| Nematodes | 17 | 162 300 | 76 681 |
| Oomycetes | 7 | 317 936 | 126 336 |
| Total | 82 | 1 529 574 | 604 969 |
| Group | No. of species | No. of ESTs and mRNAs | No. of Transcript Assemblies |
|---|---|---|---|
| Fungi | 58 | 1 049 338 | 401 952 |
| Nematodes | 17 | 162 300 | 76 681 |
| Oomycetes | 7 | 317 936 | 126 336 |
| Total | 82 | 1 529 574 | 604 969 |
A comprehensive report page is available for each TA that includes the species, taxon identifier, number of component sequences, orientation and length (Figure 2). Functional annotation is provided with the ‘top match’ of a BLAST search of the TA against the UniRef100 protein reference database, including the annotation, percent identity and percent length of the match. A component diagram indicating the individual sequence accessions used in the assembly, their orientation and relative length are depicted in the assembly diagram and tabulated in the assembly component table. Another form of annotation provided is prediction of SSRs and primers that flank the putative SSR. FASTA formatted sequence for the TA or singleton is provided to facilitate sequence-based searches by the user.
Genome and gene level tools
For whole bacterial, fungal and oomycete genomes for which the sequence and/or annotation data sets are publicly available and published, we have imported those data sets into the CPGR and provide access to the sequence and annotation through a series of interfaces that permit browsing, download and query by the user. Note that not all genomes in the warehouse are available as full data sets in the CPGR as these genomes are either still in progress or are not currently available for redistribution to third party sites such as the CPGR. For publicly available genomes, the primary method for visualizing a genome is through the Genome Browser (Figure 3), an open-source genome visualization software made available through the GMOD project (45). Currently, Genome Browser views are available for 74 annotated genomes [60 bacteria (http://cpgr.plantbiology.msu.edu/cgi-bin/gbrowse/bacteria/), 12 fungi (http://cpgr.plantbiology.msu.edu/cgi-bin/gbrowse/fungi/) and 2 oomycetes (http://cpgr.plantbiology.msu.edu/cgi-bin/gbrowse/oomycete/)]. Additional plant pathogenic bacterial, fungal, oomycete and nematode genomes will be added in the near future. The genome browsers can be accessed directly through the Genome Browser Selection tool available through the top menu bar in which a default genome is displayed for each of the phyla. Other genomes within each phylum can be viewed by selecting the GBrowse Selection Tool (http://cpgr.plantbiology.msu.edu/cgi-bin/bact_gateway.pl?page=bact_gbrowse_select; http://cpgr.plantbiology.msu.edu/cgi-bin/fungi_gateway.pl?page=fungi_gbrowse_select; http://cpgr.plantbiology.msu.edu/cgi-bin/oomycete_gateway.pl?page=oomycete_gbrowse_select) either from inside each of the three phyla level genome browsers or from the top menu bar. The CPGR genome browsers include tracks representing the loci, gene models (fungi and oomycetes only), rRNA and tRNA genes, putative SSRs, GC content and six-frame translation (Figure 3). The Scroll/Zoom tools allow the user to visualize loci throughout the entire genome at variable resolution levels. The reports/analysis tools provide for generation of decorated FASTA files and high resolution images of the genome browser. For the CPGR fungal and oomycete genome browsers, the loci are linked to a Genome Browser detail page containing coordinate, function and sequence information. Each locus in the CPGR bacterial genome browsers and each gene model in the fungal and oomycete genome browsers is hyperlinked to the respective CPGR Gene Report Page (Figure 4), which collates an array of metrics and annotation at the locus/gene model level. Functional annotation of the gene is also provided on the CPGR Gene Report Page with locus name, gene name (if available), Pfam domain matches (46), InterPro database matches (47), orthologous group membership (bacteria only) and gene name assignment. Depth of information on relatedness is provided through sequence similarity search results of the predicted protein sequence with UniRef100 (38). While the genome browser provides a mechanism to enter genomes and genes visually, the Bacterial/Fungal/Oomycete Genome Gene List page (e.g. http://cpgr.plantbiology.msu.edu/cgi-bin/bact_gateway.pl?page=all_gene_list), rRNA Gene List page (e.g. http://cpgr.plantbiology.msu.edu/cgi-bin/bact_gateway.pl?page=rna_gene_list), Pfam Domain Page (e.g. http://cpgr.plantbiology.msu.edu/cgi-bin/bact_gateway.pl?page=pfam_list) and InterPro Domain Page (e.g. http://cpgr.plantbiology.msu.edu/cgi-bin/bact_gateway.pl?page=interpro_list) provide mechanisms to obtain tabular gene sets from each genome.
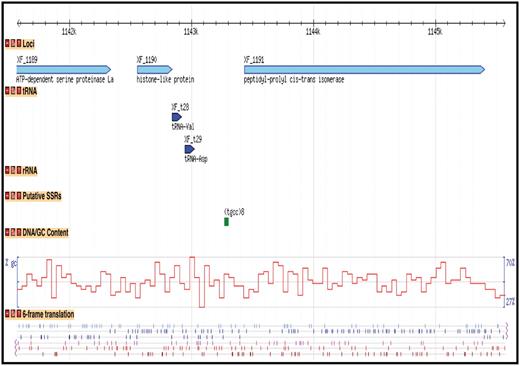
Display of bacterial genome through Genome Browser. A region of the Xylella fastidiosa 9a5c chromosome is displayed with tracks representing the loci, tRNA genes, putative Simple Sequence Repeats, GC content and 6-frame translation.
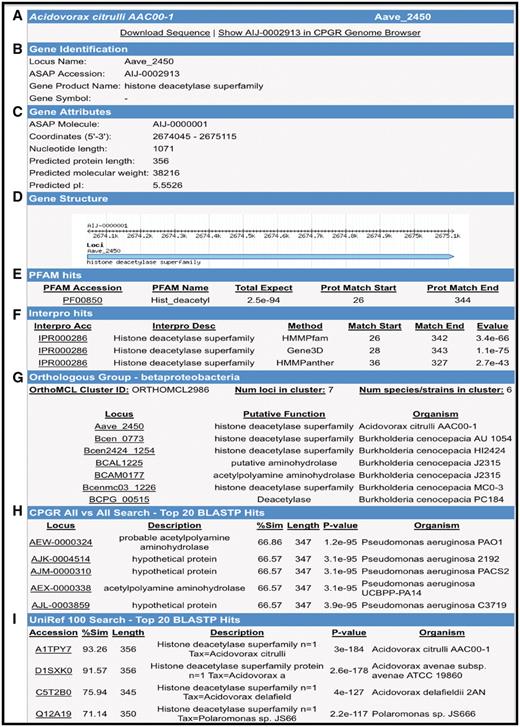
Gene Report Page for Acidovorax avenae subsp. citrulli AAC00-1 Aave_2450. (A) Hyperlinks to download gene sequence and genome browser display of the locus. (B) Locus name, functional annotation and gene name (if available). (C) Gene attributes including molecule location (chromosome, plasmid), coordinates and protein metrics. (D) Gene structure. (E) Pfam domain matches with scores. (F) InterPro hits including position of matches and E-value. (G) Orthologous groups from β-Proteobacteria clustering. (H) BLASTP search results from an all versus search of bacterial proteins within the CPGR. (I) Partial listing of UniRef100 top matches.
Precompiled whole-genome Mauve (41) alignments (http://cpgr.plantbiology.msu.edu/cpgr_asap_mauve.shtml) from within-genera groups of phytopathogenic bacteria are available for download to assist users interested in comparative genomics. The provided links download a java applet and the sequence alignment. Mauve provides a graphic view of multiple genomes with features and sequence that can be resized from an overview of the entire genome down to individual nucleotides. Features within the alignment link out to feature pages within ASAP, as well as GenBank.
For bacterial, fungal and oomycete genomes, a suite of tools is available for searching at the gene level. These include BLAST search page (e.g. http://cpgr.plantbiology.msu.edu/cpgr_bact_blast.shtml), Locus ID query page (e.g. http://cpgr.plantbiology.msu.edu/cgi-bin/bact_gateway.pl?page=locus_id_search), Putative Function search page (e.g. http://cpgr.plantbiology.msu.edu/cgi-bin/bact_gateway.pl?page=pfunc_search), Pfam domain search page (e.g. http://cpgr.plantbiology.msu.edu/cgi-bin/bact_gateway.pl?page=pfam_search) and Interpro domain search page (e.g. http://cpgr.plantbiology.msu.edu/cgi-bin/bact_gateway.pl?page=interpro_search). Due to the limited phylogenetic coverage of fungal and oomycete genomes, an orthologous group search page is available only for bacterial genomes within the CPGR (http://cpgr.plantbiology.msu.edu/cgi-bin/bact_gateway.pl?page=orthomcl_search).
Updates to each genome are tracked in the Bacteria, Fungi and Oomycete Genome Information pages. These pages display the current CPGR version of the imported genomes and annotation, the date it was released in the CPGR and the source of the original genome download. Data files for older versions such as database dumps, sequence files and BLAST databases will be maintained on the CPGR FTP server.
rDNA database
rDNA sequences, including the internal transcribed spacer (ITS) region, are widely used to develop molecular diagnostic markers for plant pathogens. To facilitate marker development for all plant pathogens, we created the rDNA database that includes sequences not only for plant pathogen taxa, but also closely related taxa and allows for stringent filtering of sequences for marker design. The rDNA sequences and associated annotations are downloaded from GenBank and stored in a MySQL relational database. A dedicated search page (http://cpgr.plantbiology.msu.edu/cgi-bin/cpgr_rdna/cpgr_rdna_db.pl) allows users to select from the nuclear and/or mitochondrial rDNA loci, the output format and then select specific loci based on genus and/or species. The current rDNA database (Version 1) contains 131 755 sequences from 17 613 species, of which 65 232 sequences are from 3760 plant pathogenic species. All sequences can be downloaded through the CPGR FTP site (ftp://ftp.plantbiology.msu.edu/pub/data/CPGR/).
Marker identification tools
SSR identification tool
To facilitate rapid discovery and testing of diagnostic SSR markers, we developed an on-line tool for plant pathologists and diagnosticians to identify candidate SSRs within a query sequence and design primers for amplification of the SSR using Primer3 (http://primer3.sourceforge.net/) (48). Users can select which type of SSR to predict both, the type of nucleotide (mononucleotide, dinucleotide, trinucleotide, etc.), as well as the number of repeats (Figure 5). As described above, SSRs have been annotated in all of the Plant Pathogen Transcript Assemblies and are viewable along with predicted primers to amplify the SSR in the Transcript Assembly Report Page (Figure 2). SSRs have also been annotated in all 74 genomes and are viewable through the Genome Browser or through the Bacteria/Fungal/Oomycete Putative SSR list query page (e.g. http://cpgr.plantbiology.msu.edu/cgi-bin/bact_gateway.pl?page=putative_ssrs).
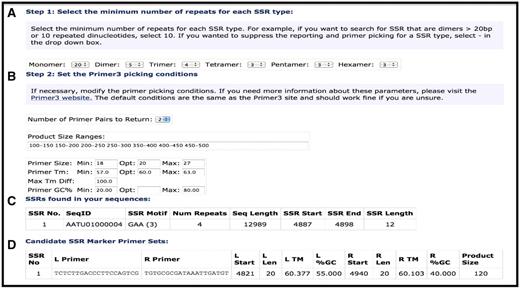
Simple Sequence Repeat search tool. (A) Selection options for SSR type and length. (B) Primer selection criteria for putative SSRs. (C) SSR Report page showing SSRs identified, motif, number of motifs and position of start/stop. (D) Predicted primers for putative SSR.
Bacterial unique loci marker search tool
Genome level sequencing of multiple taxa permits the rapid identification of unique loci that could serve as diagnostic markers. The Unique Loci Candidate Search Tool (http://cpgr.plantbiology.msu.edu/cgi-bin/bact_gateway.pl?page=unique_loci) facilitates data-mining loci of bacteria that are restricted in their phylogenetic distribution. From the Unique Loci List Query Page (http://cpgr.plantbiology.msu.edu/cgi-bin/bact_gateway.pl?page=unique_loci), the user selects a specific bacterial genome and a list of unique loci are shown with their gene symbol (if known), putative function, genome coordinates and resident molecule. The locus is hyperlinked to the Gene Report Page from which the sequence can be downloaded and primers can be picked from the loci using Primer3.
Other tools and resources
BLAST search tool
A primary method to identify sequences is through sequence-based searches such as BLAST (49). BLAST search pages are available for users to search genes, transcripts and genomes within the CPGR. These include a dedicated BLAST server for searching bacterial, fungal and oomycete loci within the CPGR using nucleotide or protein level searches (http://cpgr.plantbiology.msu.edu/cpgr_bact_blast.shtml; http://cpgr.plantbiology.msu.edu/cpgr_fungi_blast.shtml; http://cpgr.plantbiology.msu.edu/cpgr_oomycete_blast.shtml). Additional BLAST searches can be performed on the Plant Pathogen Transcript Assemblies (http://cpgr.plantbiology.msu.edu/cpgr_blast.shtml) that supports taxon selection by the user and a BLAST search against plant pathogen sequences downloaded from GenBank (http://cpgr.plantbiology.msu.edu/cpgr_pp_genbank_blast.shtml) including ESTs from dbEST, cloned genes/mRNAs/cDNAs, whole genomes and draft genome sequences and assemblies. Search results can be viewed typically in less than 1 min but are also held temporarily on a private URL for 24 h.
FTP site
The Plant Pathogen Transcript Assemblies and the collated GenBank plant pathogen sequences within the CPGR are available through FTP (ftp://ftp.plantbiology.msu.edu/pub/data/CPGR/).
Discussion
The diversity and breadth of organisms that can cause diseases on plants is vast. To facilitate access to the growing body of plant pathogen genome sequence, we have created the CPGR that serves as a portal to all publicly available plant pathogen genome sequence data and projects. Establishment of the CPGR Warehouse with accompanying metadata provides a broad, yet, detailed view of the status of plant pathogen genome sequence data. Not only are complete, publicly available data sets available, but planned and in-progress projects are collated. The CPGR supports researchers to quickly assess and obtain the genome sequence for their organisms of interest obviating the need to have either personal knowledge of the status of genomics initiatives or having to search in multiple locations for information. In addition to the Warehouse, the CPGR offers display, search and access tools to the genome sequence and annotation of 74 genomes and 82 transcriptomes. In addition, rDNA sequences are provided for 17 613 species to facilitate diagnostic marker development.
Existing web-based databases for plant pathogen genome data differ not only in terms of the number and diversity of genomes they encompass, but also in the types of data analyses supported. A number of databases support comparative genomics analysis of fungal plant pathogens; e.g. e-Fungi, COGEME and CFGP, all integrate a wide variety of fungal genomes. The Fungal Genome Initiative at the Broad Institute and the Fungal Genomics Program at the DOE Joint Genome Institute include genome sequences of select fungal plant pathogens. The Phytophthora Functional Genomics Database (PFGD) (50) and VMD are databases dedicated to plant pathogenic oomycetes including Phytophthora and Hyaloperonospora. Furthermore, there are a number of databases such as the Candida Genome Database (CGD) (51), FGDB and the Aspergillus Genome Database (AspGD) (52), which include genome sequences and other information from specific plant pathogens. Whereas these pathogen-specific databases do not support comparative analyses across a range of plant pathogen taxa, the IMG system maintained by JGI supports comparative analysis and annotation of a wide variety of microbial genomes in a comprehensive integrated context comparable with CPGR.
Genomics has resulted in fundamental improvements in the breadth and depth of our understanding of plant pathogens. For example, extensive sequencing of oomycete pathogens has revealed classes of effector molecules that modulate the host–parasite interaction (14, 53, 54). Genome-scale microarrays were used in comparative genome hybridizations to determine the core and the variable genes within the Ralstonia solanacearum genome (55). Surprisingly, of the 5074 R. solanacearum genes placed on the array, only 53% were present in all of the strains examined forming the ‘core genome’, whereas 46% were variable and present in a subset of strains. Sequencing of Fusarium graminearum, causal agent of Fusarium head blight of wheat and barley, coupled with expression profiling experiments revealed a set of 408 genes expressed exclusively during infection of barley that, based on single nucleotide polymorphism frequency were more divergent than other genes in the genome (17).
Whereas these examples show the power of genomics to advance basic research, genomics can and will have a significant role in deciphering pathogen population structure and its relationship to disease, as well as in the development of diagnostic markers for plant pathogens. For example, a number of detection methods rely on DNA-based markers where a targeted locus (loci) is amplified from the pathogen using PCR (56, 57) or detected through hybridization (58–62). Typically, these DNA-based markers can be scored in a binary fashion (present/absent), by size polymorphism, or by the kinetic nature of the amplification reaction (real-time PCR). Perhaps the most challenging aspect of developing a DNA-based marker for diagnostics is identifying unique or distinguishing loci within the target organism to provide a high resolution of detection, perhaps at the pathovar or race level. The usefulness of the CPGR as a resource was validated by Lang et al. (63) in the development of highly specific PCR-based diagnostic markers that distinguished Xanthomonas oryzae pv oryzae and Xanthomonas oryzae pv oryzicola, the causal agents of bacterial blight and bacterial leaf streak of rice, respectively. These pathovars, which are on the USDA-APHIS Select Agent list (http://www.aphis.usda.gov/programs/ag_selectagent/ag_bioterr_toxinlist.shtml), cannot easily be differentiated by morphological or physiological characteristics in culture. Using the comparative and computational resources within the CPGR, sets of unique and conserved loci were identified. These lists of candidate markers were then screened in a panel of Xanthomonas strains using PCR to validate the bioinformatics prediction of their phylogenetic distribution. Due to the availability of genome sequences from not only the target species (X. oryzae pv oryzae and X. oryzae pv oryzicola), but also other species of Xanthomonas, delineation of bona fide markers from the candidate list was straightforward, demonstrating the power of genomics, coupled with bioinformatics, to facilitate diagnostic marker development.
Next-generation sequencing methodologies, in which ultra-high-throughput sequencing capabilities are coupled with highly reduced costs (18–20, 64), enable new research directions due to the inherent paradigm-changing scale of data generation. Certainly, data handling and mining will be a large challenge and a bottleneck that needs to be addressed. However, bioinformatics solutions such as Galaxy, an open source platform for next generation sequencing computational efforts (65) are emerging to handle and process these data sets. The CPGR is already incorporating data from these methodologies and merging them with data generated from ‘first generation sequencing platforms’. Assembled genomes sequenced with next generation sequencing technologies can be readily incorporated into the ASAP and CPGR databases. In fact, 17 of the genomes obtained from ASAP were generated using next-generation sequencing technologies and were seamlessly incorporated into the CPGR. Other amenable data sets include RNA-seq data sets (66) in which mRNA is converted into cDNA and sequenced using short read next-generation platforms. Algorithms are available to perform de novo assemblies (67) of these transcriptomes that can be readily incorporated into the CPGR Transcript Assemblies database. Whereas the CPGR can currently handle the volume of plant pathogen genomes being deposited in NCBI, the pace at which genomes are being generated along with the large range in quality of genome and transcriptomes generated, will become prohibitive. As a consequence, standards for the quality of the underlying sequence for inclusion in the CPGR will need to be invoked. For example, for new genome assemblies, i.e. those without a quality reference genome, N50 contig sizes need to be sufficiently robust to permit reasonably accurate gene prediction. For transcriptome data, the quality of the underlying reads are critical to successful transcript assembly and imposition of high-quality thresholds on the sequence reads would permit more robust transcript assemblies. As these next-generation sequencing methods improve, quality criteria for reads, assemblies and annotations will stabilize and permit community-defined quality standards for genome projects that can be applied to target genomes for the CPGR.
Clearly, there is enormous potential for genomic data to shape biology, including plant pathology and the CPGR provides a portal for plant pathologists to determine the genome sequence status of their organism of interest, data mine these bacterial and eukaryotic genomes and identify candidate markers for diagnostic marker development (68).
Funding
USDA National Institute for Food and Agriculture (grant nos. 2006-55605-16645 and 2006-55605-04558 to C.R.B., J.E.L. and N.A.T.); the joint CPGR–ASAP work is funded by the USDA National Institute for Food and Agriculture (grant no. 2009-65109-05719 to C.R.B. and N.P.). Funding for open access charge: USDA National Institute for Food and Agriculture grant no. 2009-65109-05719.
Conflict of interest. None declared.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}