





Abstract
Acetogenic bacteria are imperative to environmental carbon cycling and diverse biotechnological applications, but their extensive physiological and taxonomical diversity is an impediment to systematic taxonomic studies. Acetogens are chemolithoautotrophic bacteria that perform reductive carbon fixation under anaerobic conditions through the Wood–Ljungdahl pathway (WLP)/acetyl-coenzyme A pathway. The gene-encoding formyltetrahydrofolate synthetase (FTHFS), a key enzyme of this pathway, is highly conserved and can be used as a molecular marker to probe acetogenic communities. However, there is a lack of systematic collection of FTHFS sequence data at nucleotide and protein levels. In an attempt to streamline investigations on acetogens, we developed AcetoBase - a repository and database for systematically collecting and organizing information related to FTHFS sequences. AcetoBase also provides an opportunity to submit data and obtain accession numbers, perform homology searches for sequence identification and access a customized blast database of submitted sequences. AcetoBase provides the prospect to identify potential acetogenic bacteria, based on metadata information related to genome content and the WLP, supplemented with FTHFS sequence accessions, and can be an important tool in the study of acetogenic communities. AcetoBase can be publicly accessed at https://acetobase.molbio.slu.se.
Introduction
Acetogenesis is one of the most primitive and ancient biological processes facilitating formation of organic compounds with inorganic carbon dioxide (CO2) and hydrogen (H2) by the acetyl-coenzyme A (acetyl-CoA) pathway, also referred to as the Wood–Ljungdahl pathway (WLP), a characteristic of acetogens (1–6). The importance of this process lies in its origin where there were no organic compounds to sustain life and acetate production by reduction of carbon dioxide provided enough thermodynamic potential to sustain initial chemolithoautotrophic life. Acetogens are very important in the global carbon cycle and are estimated to approximately produce 1013 kg of acetate annually in different anaerobic environments (7–10). In addition to the formation of acetate (as a main product) from inorganic carbon, acetogens are involved in degradation of organic compounds and production of secondary compounds such as ethanol, butyrate, lactate, etc. (7,11–13). Acetogens are phylogenetically highly divergent, with representatives in over 23 genera (14,15). This metabolic flexibility and phylogenetic diversity make acetogens one of the most versatile groups of microorganisms (9,16,17).
Acetogens are ubiquitous and inhabit various anaerobic environments such as anaerobic digesters, insect gut, hot springs, rumen, human gut, oilfields and lake sediments (16–19). In the recent years, acetogenesis has been gaining much attention among researchers [Figure S1 in Supplementary Information (SI)] due to its importance in anaerobic digestion, syngas fermentation, human gut physiology, production of biochemicals and synthetic biological applications etc. (7,17,20–22). However, targeted studies of the acetogenic community with modern molecular analysis tools remain limited. In the recent years, microbiome studies with the 16S rRNA gene have given an unprecedented increase in the discovery and identification of new and unculturable microbes. However, the phylogenetic divergence and metabolic versatility of acetogens makes it unfit for acetogenic community analysis (7,19,24). Decades of research on acetogenesis have revealed that it is a physiological process carried out via the WLP under specific conditions (6,14) and that development of functional markers based on the 16S rRNA gene for the acetogenic community is very arduous, if not impossible (6,25). For this reason, acetogenic community analysis by metabolic functional markers, i.e. DNA sequence motifs that can be functionally characterized in relation to certain traits (26), is preferred over 16S rRNA gene-based analysis. Fortuitously, certain genes encoding WLP enzymes, such as formyltetrahydrofolate synthetase (FTHFS) and acetyl-CoA synthetase/carbon monoxide dehydrogenase complex, are considerably conserved and good markers for probing the acetogenic community (27). The FTHFS gene is successfully being used as a metabolic functional marker for acetogenic community analysis for over two decades (7,19,22,27–31) (Figure S1). This gene is also present in syntrophic acetate oxidizing bacteria (SAOB), which are suggested to use reversed WLP for acetate oxidation (32) and has been used to target SAOB community in different methanogenic environments (33,34). However, despite extensive use of FTHFS amino acid (amino acid) and gene sequences in acetogenic community profiling, there has been no systematic sequence collection and reference database for sequence-based analysis of the acetogenic population (35). To amend this information void and to make standard resource for the study of this highly versatile and important group of bacteria, we present AcetoBase - a repository and database of FTHFS sequences for structured data collection, organization and putative taxonomic identification of sequences by homology search.
Methodology
Data retrieval and collection
Full-length FTHFS amino acid and nucleotide sequences (based on keyword, gene/protein annotations and similarity-based searches) were retrieved from the protein and nucleotide database at the National Center for Biotechnology Information (NCBI) (https://www.ncbi.nlm.nih.gov/) (36) between October and December 2018. All genome homologues of the FTHFS gene were retrieved together with genome co-ordinates and locus. Partial FTHFS clone nucleotide sequences available in the NCBI nucleotide database were also retrieved, along with metadata on their origin, isolation source, environment, temperature, etc. of original sample. Taxonomic identifiers and taxonomic lineages corresponding to amino acid and nucleotide sequences were retrieved from the NCBI taxonomy database (https://www.ncbi.nlm.nih.gov/taxonomy) (36). Whole genomic metadata were retrieved (December 2018) from the Genome Online Database (https://gold.jgi.doe.gov) (37) and the Integrated Microbial Genomes and Microbiomes database (38) under the Joint Genome Institute umbrella (https://jgi.doe.gov) (39). Based on the NCBI taxonomic identifiers of FTHFS nucleotide sequences, the corresponding 16S ribosomal RNA gene sequences were then retrieved from the SILVA database (May 2019) (https://www.arb-silva.de) (40). Additionally, whole genomes/assemblies were retrieved from the NCBI ftp site (ftp://ftp.ncbi.nlm.nih.gov) (July 2019) and genes involved in the WLP were screened based on the keyword and gene/protein annotations or The Enzyme Commission number (EC number) (Table S2) searches in genome or genomic assemblies. Non-redundant curated database for FTHFS protein and nucleotide training datasets were generated from respective AcetoBase accessions and formatted as training datasets for taxonomic assignment of next-generation sequencing (NGS) data by the AcetoScan software (Singh et al., in preparation). The trained datasets for non-redundant protein and nucleotide sequences are available at https://acetobase.molbio.slu.se/download.
Functional gene repository
AcetoBase is primarily designed to be a repository for FTHFS sequences of nucleotide, protein and clone origin (Figure 1). All sequences stored in AcetoBase are assigned with a unique identifier, which is the accession number of that entry in the database. In the AcetoBase GenBank format (41), this identifier is represented under the qualifier LOCUS and VERSION. AcetoBase sequence accessions are also prefixed with the database table to which they belong (Table 1). Registered users are permitted to submit sequences of nucleotide, protein or clone origin in a multifasta format file via the Upload menu. All uploaded sequences are screened for anomalies and automatically subjected to best frame analysis. The filtered sequence corresponding to the best open reading frame is uploaded and assigned a unique accession number based on the corresponding database table (Table 1). Sequences can be submitted as an open sequence for public access or as a personal sequence, which may not be accessed by general users without the permission of the submitter. AcetoBase LinkIn functionality facilitates direct referencing and linking to the data in database, which can also be accessed programmatically on command line interface. The description of AcetoBase GenBank accession and programmatic access via AcetoBase LinkIn is provided in SI (Data D1–D5). Upon blast query submission, the user sequence data are formatted as a custom blast database (42), which can be used directly to carry out homology analysis. AcetoBase is linked to NCBI via the LinkOut project for the cross platform data sharing and integration (43).
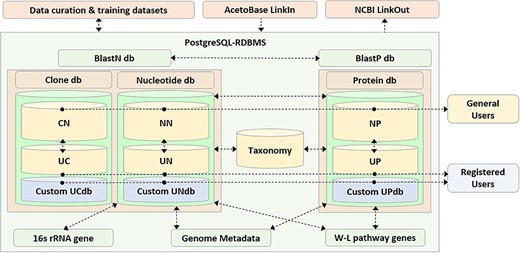
Schematic representation of the AcetoBase repository and database. The nucleotide database (db) table consists of reference nucleotide (NN) table and user nucleotide (UN) table, the clone database table consists of reference clone (CN) table and user clone (UC) table and the protein database table consists of reference protein (NP) table and user protein (UP) table. General users have access to all sequences in the databases except for those that are not yet public, while registered users also have access to the custom database generated with their own clone (UCdb), nucleotide (UNdb) and protein (UPdb) sequences. W-L, Wood–Ljungdahl.
Synopsis of the AcetoBase accession number format, with prefix denoting corresponding database, the ownership of the sequence and the status of the sequence
| Prefix & accession number | AcetoBase databases | Owner/maintainer | Status |
|---|---|---|---|
| NN_0000012345 | Nucleotide database | Publication author/admin | Reference |
| NP_0000012345 | Protein database | Publication author/admin | Reference |
| CN_0000012345 | Clone database | Publication author/admin | Reference |
| UN_0000012345 | User Nucleotide database | User name, email and affiliation | New |
| UP_0000012345 | User Protein database | User name, email and affiliation | New |
| UC_0000012345 | User clone database | User name, email and affiliation | New |
| Prefix & accession number | AcetoBase databases | Owner/maintainer | Status |
|---|---|---|---|
| NN_0000012345 | Nucleotide database | Publication author/admin | Reference |
| NP_0000012345 | Protein database | Publication author/admin | Reference |
| CN_0000012345 | Clone database | Publication author/admin | Reference |
| UN_0000012345 | User Nucleotide database | User name, email and affiliation | New |
| UP_0000012345 | User Protein database | User name, email and affiliation | New |
| UC_0000012345 | User clone database | User name, email and affiliation | New |
Synopsis of the AcetoBase accession number format, with prefix denoting corresponding database, the ownership of the sequence and the status of the sequence
| Prefix & accession number | AcetoBase databases | Owner/maintainer | Status |
|---|---|---|---|
| NN_0000012345 | Nucleotide database | Publication author/admin | Reference |
| NP_0000012345 | Protein database | Publication author/admin | Reference |
| CN_0000012345 | Clone database | Publication author/admin | Reference |
| UN_0000012345 | User Nucleotide database | User name, email and affiliation | New |
| UP_0000012345 | User Protein database | User name, email and affiliation | New |
| UC_0000012345 | User clone database | User name, email and affiliation | New |
| Prefix & accession number | AcetoBase databases | Owner/maintainer | Status |
|---|---|---|---|
| NN_0000012345 | Nucleotide database | Publication author/admin | Reference |
| NP_0000012345 | Protein database | Publication author/admin | Reference |
| CN_0000012345 | Clone database | Publication author/admin | Reference |
| UN_0000012345 | User Nucleotide database | User name, email and affiliation | New |
| UP_0000012345 | User Protein database | User name, email and affiliation | New |
| UC_0000012345 | User clone database | User name, email and affiliation | New |
Structure of database
AcetoBase (Figure 1) is created in the PostgresSQL relational database management system (RDBMS) version 10.9 (44), running on Linux OS (Ubuntu 10.9-0ubuntu0.18.04.1) on x86_64-pc-linux-gnu, compiled by gcc (Ubuntu 7.4.0-1ubuntu1~18.04.1) 7.4.0, 64-bit (45). The software code for the database is written in Python3 version 3.6.7 (46), Biopython version 1.74 (47), Nginx version 1.17.0 (48), Flask web framework version 1.0.2 (49) and Jinja2 version 2.7 (50). Phylotree.js is used to render phylogenetic trees on the web interface (51).
In AcetoBase, the term nucleotide refers to gene sequences originating from whole genome sequencing, amplicon sequencing or metagenomics sequencing experiments. Thus, nucleotide is synonymous with gene in the AcetoBase context. Sequences originating specifically from cloning experiments are termed clone and included in the clone dataset. AcetoBase in its initial release contains approximately 13 000 full-length FTHFS nucleotide sequences, 18 000 full-length FTHFS amino acid sequences and 3000 FTHFS clone sequences spanning a total of 7928 NCBI taxonomic identifiers (taxid/taxon) (52). A total of 6582 whole genome/genomic assemblies from 7928 taxids were successfully retrieved to screen for presence or absence of genes (based on genome annotations) relating to the WLP. Approximately 5560/7928 taxids contain the whole genome/sequencing project metadata information, and 2010/7928 accessions are supplemented with 16S ribosomal RNA gene sequences.
Four relational database tables were created, for the nucleotide, protein, taxonomy and clone datasets, and further linked to the sub-dataset tables containing the genome metadata, WLP genes identifiers and 16S rRNA gene sequence information (Figure 1). The information stored in a relational database was then rendered in GenBank flat file format (53). A few changes were made to the standard GenBank format for standardization and homogeneity of records, and some extra flags (described below) were introduced (41). For AcetoBase nucleotide accessions, the metadata and 16S rRNA sequence information are linked to AcetoBase records via the qualifier /db_xref = taxon: and /16S rRNA gene =, respectively. Flag /WLP_genes = was introduced to link the AcetoBase entry to the metadata information regarding the presence of genes involved in the WLP to the corresponding taxid. Similar to nucleotide accessions, protein accessions were also rendered in GenBank format along with the qualifier /coded_by =, which contains the genome coordinates of the nucleotide sequence. For the FTHFS clone sequence AcetoBase records, some new qualifiers that might be relevant to the user were introduced. These qualifiers are /putative_taxonomy = , /clone_env, /temp, /NH4_N and /pH. Qualifiers are intended to collect more information about the clone environment, temperature, concentration of ammonium-nitrogen, pH and other important details in comment box, wherever possible for the new sequences.
Phylogenetic inference of FTHFS datasets
Phylogenetic trees for the non-redundant FTHFS protein, nucleotide and clone datasets were computed using IQ-tree (54) on the SLUBI computing cluster in Uppsala, running CentOS Linux release 7.1.1503 with module handling by Modules based on Lua: Version 6.0.1 (55). A maximum likelihood tree was constructed with 1000 ultrafast bootstrap (UFBoot2) (56), 1000 SH-like approximate likelihood ratio test (SH-alrt) (57), with a scalable parallel random number generator value (SPRNG) of 12 (seed) for each of the datasets. The WAG+GAMMA4 model (58,59) was used for the protein and translated clone datasets, and the GTR + GAMMA4 model (59,60) was used for the nucleotide dataset. Clustering of the homologous sequences in phylogenetic trees was performed in Cluster Picker with default parameters (61) and TreeCluster (62) with a distance threshold value of 1. The resulting IQ-tree phylogenetic trees of FTHFS protein, nucleotide and translated clone datasets were rendered in an interactive web-interface on AcetoBase using phylotree.js plugin.
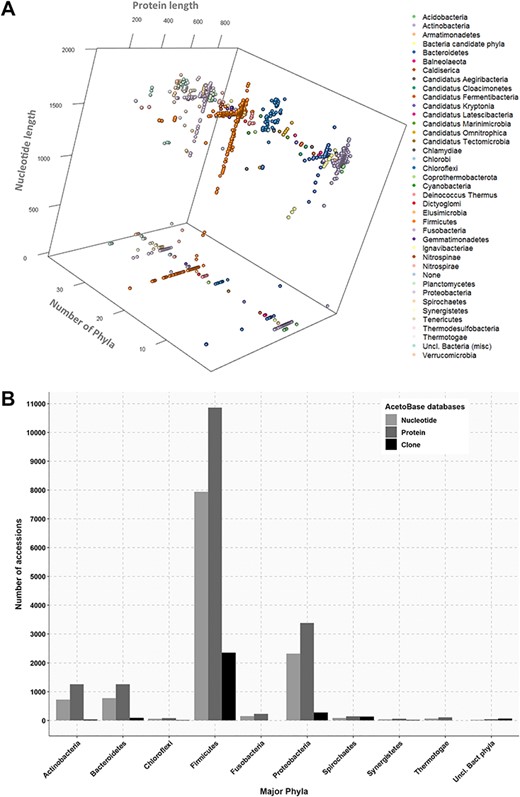
(A) Three-dimensional projection of nucleotide and protein sequence length according to phylum. Length of protein sequences, length of nucleotide sequences and number of phyla are shown on the x-axis, y-axis and z-axis, respectively. Protein sequences without a corresponding nucleotide sequence in the database are indicated on the z-plane. (B) Phylum affiliations of nucleotide, protein and clone accessions in AcetoBase. Phyla containing less than 20 accessions are not shown.
Taxonomy prediction for FTHFS clone dataset
An additional qualifier, /putative_taxonomy = , was introduced in AcetoBase GenBank format (41) to give an indication of the possible taxonomic affiliation of the sequence, as most of the clones in public databases are submitted as uncultured bacteria. Putative taxonomy was predicted by the SINTAX algorithm (63), using the curated reference AcetoBase protein dataset. The SINTAX algorithm was standardized for use with FTHFS protein sequences, since to our knowledge it has not been used previously to elucidate the percentage identity threshold at various taxonomic levels for this specific protein sequence. To standardize SINTAX taxonomic prediction, 145 full-length FTHFS protein sequences with known taxonomy were randomly selected from AcetoBase. Datasets of different amino acid length (490, 420, 350, 280, 210, 140, 70 and 35) were prepared by trimming full-length protein sequences. These datasets were used to predict the taxonomic affiliations by SINTAX algorithm against the protein training dataset (available at https://acetobase.molbio.slu.se/download) in USEARCH software version 10 (64).
Percentage identity thresholds for different taxonomic levels were also calculated by similarity matrix analysis of protein and nucleotide sequences of known taxonomy. The class Clostridia in the phylum Firmicutes was selected for the analysis, as it is one of the most abundant and divergent classes in overall AcetoBase accessions (Figure 2B). To configure minimum percentage similarity cut-off for different taxonomic levels, 310, 262, 60 and 36 sequences of the same Class, Order, Family and Genus level, respectively, were selected (Figure 3). Selected full-length nucleotide sequences of the class Clostridia were aligned to the clone sequences generated with the primer set designed by Müller et al. (33) and trimmed to 588 base pairs (bp) corresponding to the clone sequences. Similarly, full-length protein sequences were aligned to the translated clone sequences and trimmed to 196 aa. Thereafter, full-length nucleotide and protein sequences were compared against the trimmed nucleotide and trimmed protein sequences, respectively (Figure 3).
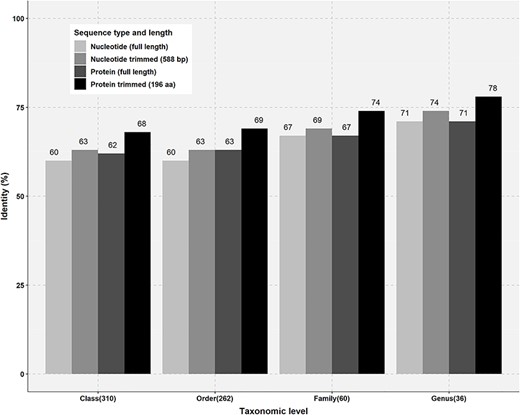
Percentage identity thresholds of FTHFS sequences at different taxonomic levels. Full-length and trimmed sequences of known taxonomy for class (310), order (262), family (60) and genus (36) level were compared with clone sequences of 588 bp and translated sequences of 196 aa.
Prediction of homoacetogenic metabolism
Prediction of homoacetogenic metabolism was attempted from FTHFS protein sequences with the signature amino acid residues in the acetogen-specific sequences, as proposed by Henderson et al. (65). In this study, 40 acetogens with homoacetogenic metabolism were selected from lists of species given in the literature (14,19) and 53 full-length FTHFS amino acid sequences (from 40 acetogens) present in AcetoBase were aligned and curated. Multiple sequence alignment was performed separately by MAFFT (version 7) (66) and MUSCLE (version 3) (67), with 1000 iterations. The multiple sequence alignment resulting from MAFFT was employed for Hidden Markov Model (HMM) profile generation using the hmmbuild command with --amino flag and a sliding window length of four amino acid residues with random seed value of 500 in HMMER3 (68). The profile HMM was used to screen the true acetogen FTHFS sequence from the protein training dataset, using strict screening with hmmsearch command --max flag (maximum sensitivity) and a minimum threshold of 94%.
Results
Size distribution of AcetoBase FTHFS sequence accessions
In addition to the wide phylogenetic variation among acetogens, there is also a huge variation in size of FTHFS protein and nucleotide sequences. To visualize the extent of size variations among these sequence datasets, a three-dimensional projection was performed for FTHFS protein and nucleotide sequences sizes (at phylum level) as presented in Figure 2A. The size distribution for the nucleotide dataset ranged from a minimum of 105 bp to a maximum of 1962 bp (mean 1169 bp). The size distribution for the protein sequences ranged from a minimum of 34 aa to a maximum of 935 aa (mean 556 aa). AcetoBase FTHFS clones sequences generated from different primer sets (9,22,29,66,67) had a size distribution ranging from 201 bp to 1128 bp (data not shown). Approximately 5000 FTHFS amino acid sequences (originated from the NCBI protein cluster analysis) (71) present in the AcetoBase protein database do not have corresponding nucleotide coordinates in the genome (71) and thus lack link to the AcetoBase nucleotide database (Figure 2A).
Cut-off threshold for different phylogenetic levels in FTHFS sequence similarity analysis
For confident determination of the taxonomy based on sequence similarity at different taxonomic levels, the percentage threshold is very important parameter. This parameter can help in the sequence classification of the new FTHFS sequences for every taxonomic level. In our analysis, based on the results from SINTAX standardization and percentage similarity analysis, the minimum percentage similarity thresholds were enumerated. The thresholds for Phylum, Class, Order, Family and Genus level were determined to be 60%, 68%, 69%, 74% and 78%, respectively (Figure 3). Taxonomic prediction with SINTAX using the protein training dataset emphasized that the similarity percentage could be considered for evaluation of any particular query sequence at different taxonomic levels. We successfully annotated FTHFS clone sequences with full taxonomic lineage under the /putative_taxonomy flag in AcetoBase and propose that annotations are correct at the threshold level as presented in Figure 3.
Analysis of FTHFS sequence phylogeny
Phylogenetic inference of AcetoBase FTHFS protein, nucleotide and clone datasets performed in this study are the most comprehensive phylogenetic trees for FTHFS sequences so far. In the phylogenetic tree (maximum likelihood) construction the computation time for the 1000 bootstrap tree was 2416, 2123 and 1308 h for the FTHFS protein, FTHFS nucleotide and FTHFS clone datasets, respectively. However, the UFBoot analysis did not converge and the phylogenetic inferences were thus terminated after 1000 iterations. Cluster analysis with Cluster Picker was unsuccessful, since no clusters were detected in any of the trees. Further analysis of clustering in phylogenetic trees was performed using TreeCluster, which revealed 197, 403 and 70 clusters in protein, nucleotide and clone trees, respectively. Acetogens appeared to be clustered in different clusters in the protein and nucleotide trees, and no meaningful acetogenesis-specific clusters were observed. A pattern of clustering was observed in the clone tree and most clusters appeared to have the same experimental origin (primers used) and taxonomic affiliations. Phylogenetic trees for protein, nucleotide and clone datasets can be accessed at the following links https://acetobase.molbio.slu.se/phylo/prot, https://acetobase.molbio.slu.se/phylo/nuc and https://acetobase.molbio.slu.se/phylo/clone, respectively.
Prediction of homoacetogens in the protein training dataset
Multiple sequence alignments for 53 FTHFS amino acid sequences performed by MAFFT and MUSCLE were compared to check for any anomalies in the conserved amino acid residues. It was found that the alignments differed only in position 479 and 480 for amino acid residue S and L, respectively, compared with the Moorella thermoacetica FTHFS amino acid sequence. The alignment resulting from MAFFT was in accordance with the amino acid residues (40 aa residue between position 181–484 of M. thermoacetica FTHFS amino acid sequence) (Figure 4) suggested by Henderson et al. (65), and therefore MAFFT alignment was used for HMM profiling. It is worth mentioning that we did not find the sequence conservations as suggested by Henderson et al. (65) in our alignment of 53 sequences from 40 acetogens (Figure 4; Table S1). Furthermore, screening of the AcetoBase protein training dataset with profile HMM resulted in 327 sequences above the 94% threshold. The E-value ranged from 0 to 8e-295 and the score from 1039 to 977.5, and the average bias was 5.46. Of the 327 sequences, 89 (excluding profile HMM sequences) were found to have an E value of 0. The 327 sequences were taxonomically assigned to 144 species. Surprisingly, it was noted that out of 53 sequences from known acetogens in profile HMM, only 21 sequences had an E value of 0, while 15 sequences had an E value of 2.8e-304–8e-295 and 17 sequences were not reported in the final result list when the cut-off was set to 94%.
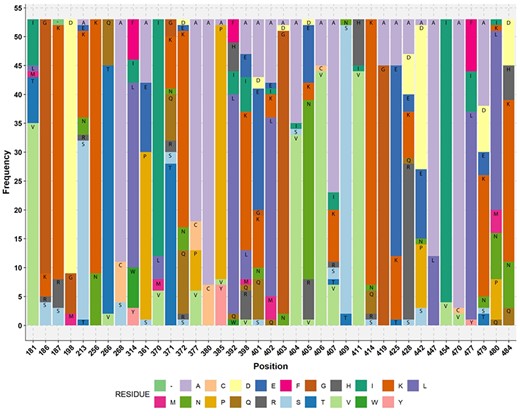
Stacked bar plot showing the amino acid residue of acetogens corresponding to the reference M. thermoacetica FTHFS aa sequence. The aa residue position of M. thermoacetica is shown on the x-axis and the frequency of the aa residue in the multiple alignment of 53 sequences of known acetogens (M. thermoacetica included) is shown on the y-axis. Minus sign (−) denotes absence of any amino acid residue.
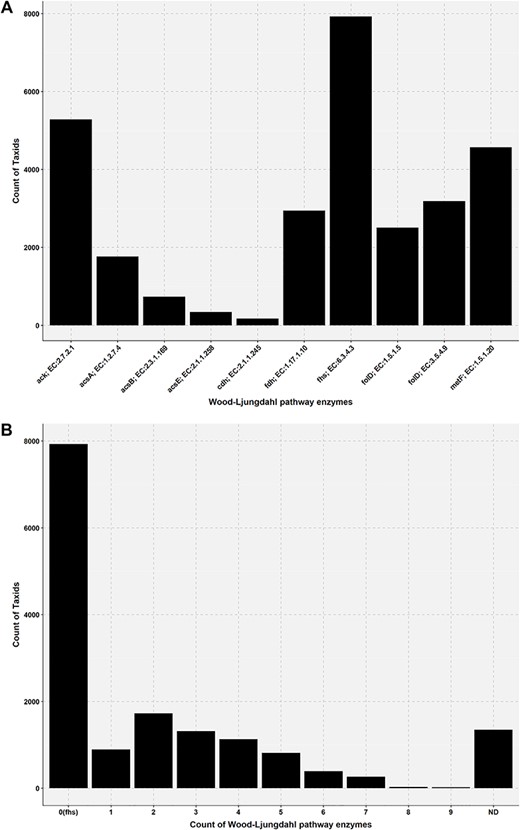
Bar plot showing the presence of Wood–Ljungdahl pathway enzymes in the genome of AcetoBas accessions. (A) Count of taxids for the Wood–Ljungdahl pathway enzymes in the genome of AcetoBase accessions (ND, not determined). (B) Count of taxids for the presence of particular enzymes in the Wood–Ljungdahl pathway.
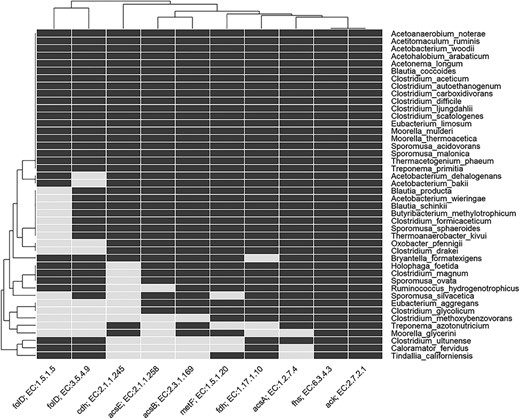
Heatmap indicating the presence and absence of enzymes involved in the Wood–Ljungdahl pathway in genome of known acetogens. The rows show the named acetogens, and the columns show the EC number and gene name for the enzymes. For details of enzyme EC number, etc., see Table S2 in Supplementary Information.
Discussion
AcetoBase is the first repository/database to systematically organize molecular marker information on acetogenic bacteria at nucleotide/gene, protein and clone level. Through AcetoBase, a majority of FTHFS sequences can be linked to WLP, whole genomics metadata and 16S rRNA gene sequences. AcetoBase is a public repository and database enabling FTHFS sequence homology analysis against a huge collection of curated datasets and custom databases for user-specific sequences. In the present study, we also identified taxonomic thresholds under which homology analyzed for different sequence types and lengths can be highly probable for different taxonomic levels (Figure 3). In our analysis, we found that these individual taxonomic level similarity thresholds were superior to a common similarity search by the blast algorithm, which results in percentage similarity prediction of taxonomic leaves, rather than taxonomic prediction of taxonomic nodes and leaves. Standardization of FTHFS sequence similarity for various taxonomic levels also suggested that taxonomic predictions for the sequences presented in the clone database are credible within the thresholds identified in this study.
Although FTHFS is a functional marker for acetogens, it can still be present in the genome of non-acetogens (6). Interestingly, some SAOB can be acetogens (Thermacetogenium phaeum) or non-acetogens (Thermotoga lettingae) and harbor FTHFS gene (10,20,32). The WLP or some genes related to it, especially FTHFS gene, are also present in sulfate-reducing bacteria (7) and Archaea, such as methanogens (72,73). Moreover, the FTHFS gene is also present in bacteria not possessing the WLP, such as Lactobacillus, where it serves the function of activation of formate to formate-tetrahydrofolate for anabolic purposes (74,75). AcetoBase contains sequences from all above-mentioned bacterial groups, however, the Archaeal FTHFS sequence has not (yet) been incorporated.
The phylogenetic inference performed in this study is the most descriptive analysis of FTHFS protein, nucleotide or clone sequences to date. The clustering pattern in the tree generated by protein sequences was not similar to that in the nucleotide sequence tree. However, some acetogen sequences appeared to cluster together in both trees, although without any particular pattern. It can be debated whether this clustering was due to sequence conservation of acetogens or simply reflected the fact that most acetogens belong to the same or a closely related genus. Interestingly, upon further analysis of clusters in the clone sequence tree and cross-referencing with AcetoBase clone metadata information, it was observed that these sequences clustered mainly because they were generated by the same primers or were of approximately the same size. However, when the AcetoBase putative taxonomy for clone sequences was taken into consideration, the reason of clustering appeared to be the taxonomic origin of sequences. For example, a well-studied set of clones (139 sequences) from termite gut (68–74) clustered together. The predicted taxonomy in our analysis indicated that these sequences are Treponema primitia clones, which is probably the primary reason for their clustering. The analysis of another clone dataset from anaerobic digestion systems revealed that 233 clone sequences from anaerobic digester environments (30,61,75–80) were clustered into 34 different clusters having 86 different putative genera. Therefore, we can confidently say that clustering of sequences in FTHFS protein and nucleotide trees cannot be interpreted as an indication of bacteria with acetogenic metabolism.
In our analysis of FTHFS amino acid sequences from known acetogens (Figure 4), regardless of the alignment algorithm used, acetogen amino acid residues appeared not to have position conservation corresponding to FTHFS amino acid residue positions of reference M. thermoacetica as suggested by Henderson et al. (65). Additionally, HMM profile scanning conflicted with the 94% cut-off threshold, as it excluded some acetogens from the HMM scanning results. Thus, we propose that Homoacetogen Similarity score cannot be used as a criterion to decide whether an FTHFS amino acid sequence originates from an acetogen or not and its limitations has already been pointed out by the authors (65). Moreover, we observed that the selection of FTHFS sequences can greatly influence the scoring result in HMM screening, and thus such analysis can be biased or selective and not absolute in screening for potential acetogenic candidates. The phylogenetic inference from FTHFS sequences and profile HMM analysis revealed that, although the FTHFS sequence is highly conserved in the WLP, it cannot be used to predict physiological capability for acetogenesis (6,25).
Acetogenesis is a physiological trait and the concept of acetogen/homoacetogen is very complex (6,24,88). A number of acetogens have been shown to have a metabolism that is non-acetogenic under certain conditions (14,16). For instance, the acetogen M. thermoacetica does not have an acetogenic metabolism when grown in nitrate-rich medium, while the acetogen Acetobacterium woodii does not have an acetogenic metabolism when using phenyl acrylates and lignin derivatives for growth (14). This ambiguity has also been pointed out several times in scientific literature and according to Müller and Frerichs (14), the term homoacetogen should not be applied to a group of organisms but rather the term homoacetogenesis, describing microbial activity under specific conditions that support homoacetogenic metabolism. To get an indication of acetogenic potential of any candidate bacteria, presence of the WLP must be considered. In the present analysis, we found that only a few genomes have all the enzymes of WLP (Figures 5 and 6; Table S2). This might be due to the absence of WLP genes in the genome or the genes have not been found in our current analysis. In AcetoBase, we have attempted to supplement the FTHFS sequences with enzymes related to the WLP for that particular accession, and we recommend that presence/absence of these enzymes might be considered when deciding on potential acetogenic candidates. In this regard, AcetoBase can be considered an important tool, apart from a repository and a database, in the prediction of potential acetogens. However, only physiological characterization for acetogenic metabolism can verify whether the candidate is an acetogen.
Future perspectives
Since NGS technologies are being developed at an exponential rate, it is highly likely that FTHFS will be used extensively in future for high-throughput analyses of acetogenic populations in natural and constructed environments and in human and animal gut. Analysis of high-throughput sequencing data requires a specific software platform and efficient computing infrastructure (89). Such computational capacity and expertise in data analysis are not easily accessible to many researchers working directly or indirectly on acetogenesis and acetogenic bacteria. A future step in the development of AcetoBase will be incorporation of web-based pipeline for analysis and visualization of FTHFS high throughput NGS data.
Availability
AcetoBase is available for public use as a functional gene repository and database and can be accessed at https://acetobase.molbio.slu.se/. The database also hosts the reference training datasets for the nucleotide and protein sequences.
Acknowledgements
We would like to thank Prof. Erik Bongcam-Rudloff and Swedish University of Agricultural Sciences Bioinformatics Infrastructure (SLUBI) for providing the computational platform required for large multiple sequence alignments, phylogenetic tree generation and all other complex bioinformatics analysis. This work was funded and supported by the Swedish Energy Agency (project no. 2014-000725), Västra Götaland Region (project no. MN 2016-00077) and Interreg Europe (project Biogas2020).
Conflict of interest. None declared.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}